Гемопоэз и лейкозы у пациентов с синдромом Дауна |
1. Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355(9199):165-9
2. Zipursky A. Susceptibility to leukemia and resistance to solid tumors in Down syndrome. Pediatr Res. 2000;47(6):704
3. Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113(12): 2619-28
4. Roy A, Roberts I, Norton A, Vyas P. Acute megakaryoblastic leukaemia (AMKL) and transient myeloproliferative disorder (TMD) in Down syndrome: a multi-step model of myeloid leukaemogenesis.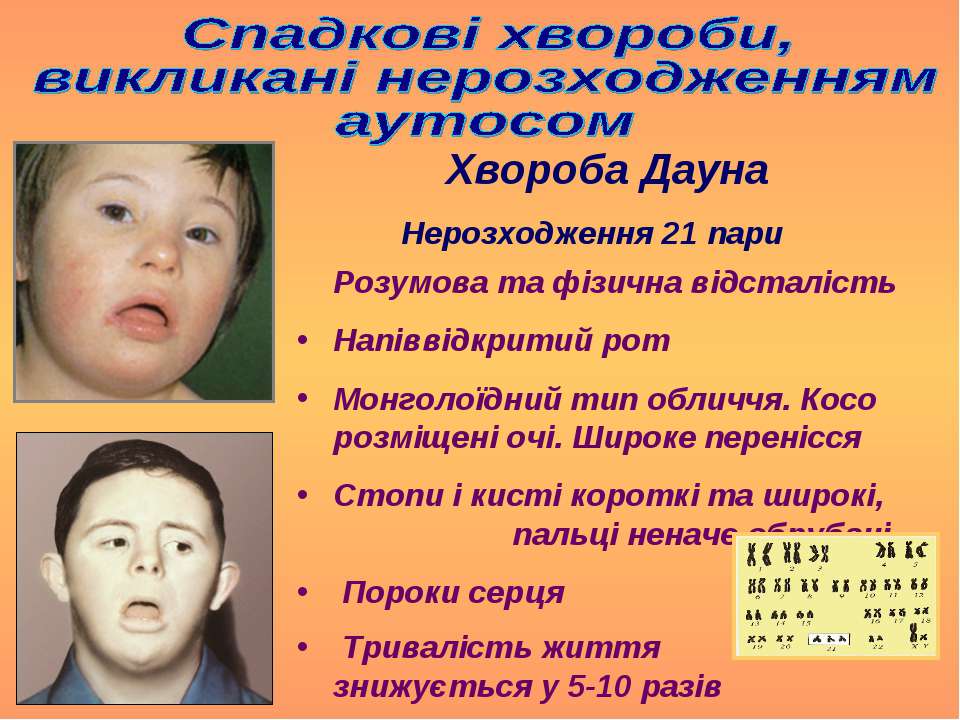 Br J Haematol. 2009;147(1):3-12
Br J Haematol. 2009;147(1):3-12
5. Izraeli S, Vora A, Zwaan CM, Whitlock J. How I treat ALL in Down’s syndrome: pathobiology and management. Blood. 2014;123(1):35-40
6. Zwaan MC, Reinhardt D, Hitzler J, Vyas P. Acute leukemias in children with Down syndrome. Pediatr Clin North Am. 2008;55(1):53-70
7. Taga T, Saito AM, Kudo K, Tomizawa D, Terui K, Moritake H, et al. Clinical characteristics and outcome of refractory/relapsed myeloid leukemia in children with Down syndrome. Blood. 2012;120(9):1810-5
8. O’Brien MM, Cao X, Pounds S, Dahl GV, Raimondi SC, Lacayo NJ, et al. Prognostic features in acute megakaryoblastic leukemia in children without Down syndrome: a report from the AML02 multicenter trial and the Children’s Oncology Group Study POG 9421. Leukemia. 2013;27(3):731-4
Leukemia. 2013;27(3):731-4
9. Buitenkamp TD, Izraeli S, Zimmermann M., Forestier E, Heerema NA, van den Heuvel-Eibrink MM, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123(1):70-7
10. Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalová K, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111(3):1575-83
11. Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372(9648):1484-92
12. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243-6
Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243-6
13. Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688-98
14. Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115(5):1006-17
15. Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, et al. Gain-of- function mutations in interleukin-7 receptor- (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208(5):901-8
Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, et al. Gain-of- function mutations in interleukin-7 receptor- (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208(5):901-8
16. Tal N, Shochat C, Geron I, Bercovich D, Izraeli S. Interleukin 7 and thymic stromal lymphopoietin: from immunity to leukemia. Cell Mol Life Sci. 2014;71(3):365-78
17. Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17(2):277-82
18. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes.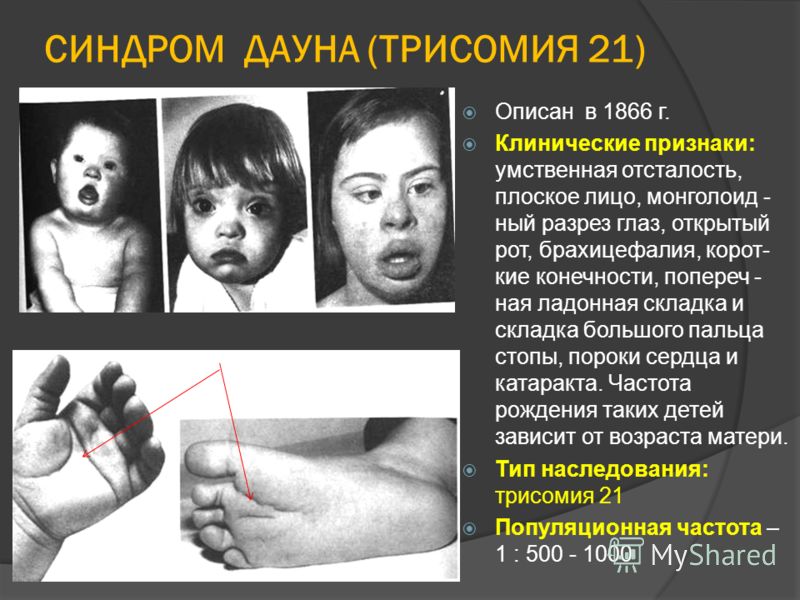 Blood. 2009;114(5):937-51
Blood. 2009;114(5):937-51
19. Langebrake C, Creutzig U, Reinhardt D. Immunophenotype of Down syndrome acute myeloid leukemia and transient myeloproliferative disease differs signifi- cantly from other diseases with morphologically identical or similar blasts. Klin Pädiatr. 2005;217(3):126-34
20. Ge Y, Jensen TL, Stout ML, Flatley RM, Grohar PJ, Ravindranath Y, et al. The role of cytidine deaminase and GATA1 mutations in the increased cytosine arabinoside sensitivity of Down syndrome myeloblasts and leukemia cell lines. Cancer Res. 2004;64(2):728-35
21. Ge Y, Stout ML, Tatman DA, Jensen TL, Buck S, Thomas RL, et al. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005;97(3):226-31
22. Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, et al. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114(13):2744-52
Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, et al. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114(13):2744-52
23. Nikolaev SI, Santoni F, Vannier A, Falconnet E, Giarin E, Basso G, et al. Exome sequencing identifies putative drivers of progression of transient myeloproliferative disorder to AMKL in infants with Down syndrome. Blood. 2013;122(4):554-61
24. Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013;45(11):1293-9
25. Greaves M. Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol. 2003;7(3):233-45
26. Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, et al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111(6):2991-8
Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, et al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111(6):2991-8
27. Zipursky A. Transient leukaemia—a benign form of leukaemia in newborn infants with trisomy 21. Br J Haematol. 2003;120(6):930-8
28. Gamis AS, Alonzo TA, Gerbing RB, Hilden JM, Sorrell AD, Sharma M, et al. Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children’s Oncology Group Study A2971. Blood. 2011;118(26):6752-9
29. Roberts I, Alford K, Hall G, Juban G, Richmond H, Norton A, et al. GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia.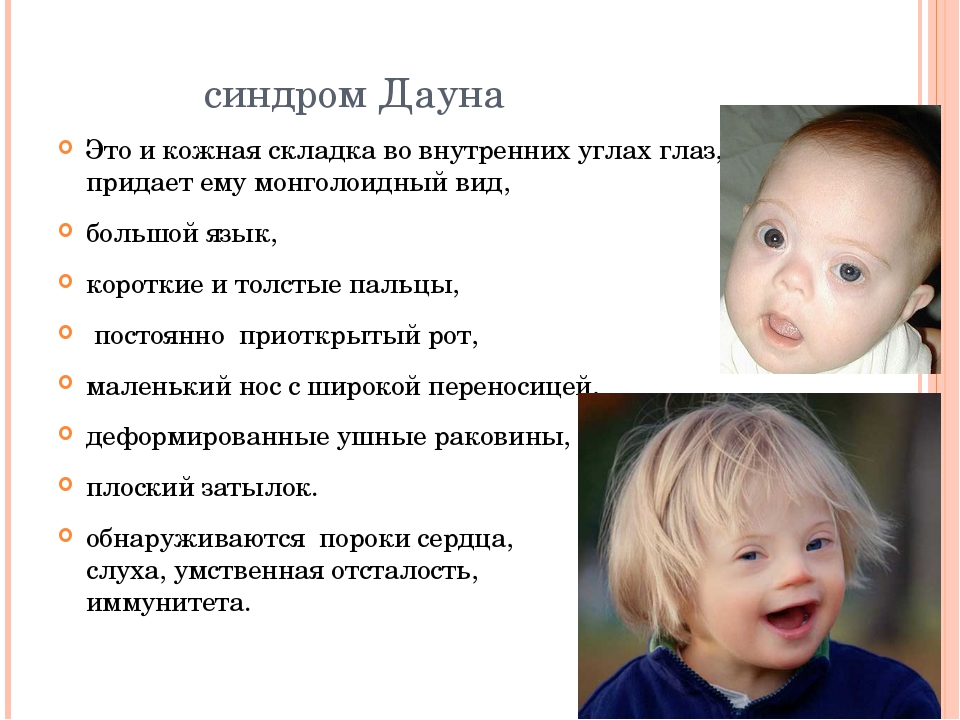 Blood. 2013;122(24): 3908-17
Blood. 2013;122(24): 3908-17
30. Groet J, McElwaine S, Spinelli M, Rinaldi A, Burtscher I, Mulligan C, et al. Acquired mutations in GATA1 in neonates with Down’s syndrome with transient myeloid disorder. Lancet. 2003;361(9369):1617-20
31. Hitzler JK, Cheung J, Li Y, Scherer SW, Zipursky A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood. 2003;101(11):4301-4
32. Rainis L, Bercovich D, Strehl S, Teigler-Schlegel A, Stark B, Trka J, et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003;102(3):981-6
33. Ahmed M, Sternberg A, Hall G, Thomas A, Smith O, O’Marcaigh A, et al. Natural history of GATA1 mutations in Down syndrome. Blood. 2004;103(7):2480-9
Natural history of GATA1 mutations in Down syndrome. Blood. 2004;103(7):2480-9
34. Pine SR, Guo Q, Yin C, Jayabose S, Druschel CM, Sandoval C. Incidence and clinical implications of GATA1 mutations in newborns with Down syndrome. Blood. 2007;110(6):2128-3
35. Heald B, Hilden JM, Zbuk K, Norton A, Vyas P, Theil KS, Eng C. Severe TMD/AMKL with GATA1 mutation in a stillborn fetus with Down syndrome. Nat Clin Pract Oncol. 2007;4(7):433-8
36. Massey GV, Zipursky A, Chang MN, Doyle JJ, Nasim S, Taub JW, et al. A pros- pective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children’s Oncology Group (COG) study POG-9481. Blood. 2006;107(12):4606-13
37. Maroz A, Stachorski L, Emmrich S, Reinhardt K, Xu J, Shao Z, et al. GATA1s induces hyperproliferation of eosinophil precursors in Down syndrome transient leukemia. Leukemia. 2014;28(6):1259-70
Maroz A, Stachorski L, Emmrich S, Reinhardt K, Xu J, Shao Z, et al. GATA1s induces hyperproliferation of eosinophil precursors in Down syndrome transient leukemia. Leukemia. 2014;28(6):1259-70
38. Lange BJ, Kobrinsky N, Barnard DR, Arthur DC, Buckley JD, Howells WB, et al. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children’s Cancer Group Studies 2861 and 2891. Blood. 1998;91(2):608-15
39. Hasle H, Abrahamsson J, Arola M, Karow A, O’Marcaigh A, Reinhardt D, et al. Myeloid leukemia in children 4 years or older with Down syndrome often lacks GATA1 mutation and cytogenetics and risk of relapse are more akin to sporadic AML. Leukemia. 2008;22(7):1428-30
40. Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32(1):148-52
Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32(1):148-52
41. Xu G, Nagano M, Kanezaki R, Toki T, Hayashi Y, Taketani T, et al. Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood. 2003;102(8):2960-8
42. Alford KA, Reinhardt K, Garnett C, Norton A, Böhmer K, von Neuhoff C, et al. Analysis of GATA1 mutations in Down syndrome transient myeloproliferative disorder and myeloid leukemia. Blood. 2011;118(8):2222-38
43. Taub JW, Mundschau G, Ge Y, Poulik JM, Qureshi F, Jensen T, et al. Prenatal origin of GATA1 mutations may be an initiating step in the development of megakaryocytic leukemia in Down syndrome. Blood. 2004;104(5):1588-9
44. Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38(7):807-12
Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38(7):807-12
45. Ganmore I, Smooha G, Izraeli S. Constitutional aneuploidy and cancer predisposition. Hum Mol Genet. 2009;18(R1):R84-93
46. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149(5):179-93
47. Nižetić D, Groet J. Tumorigenesis in Down syndrome: big lessons from a small chromosome. Nat Rev Cancer. 2012;12(10):721-32
48. Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3):266-70
Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3):266-70
49. Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122(7):2439-43
50. Klar J, Khalfallah A, Arzoo PS, Gazda HT, Dahl N. Recurrent GATA1 mutations in Diamond-Blackfan anaemia. Br J Haematol. 2014;166(6):949-51
51. Salek-Ardakani S, Smooha G, de Boer J, Sebire NJ, Morrow M, Rainis L, et al. ERG is a megakaryocytic oncogene. Cancer Res. 2009;69(11):4665-73
52. Toki T, Kanezaki R, Kobayashi E, Kaneko H, Suzuki M, Wang R, et al. Naturally occurring oncogenic GATA1 mutants with internal deletions in transient abnormal myelopoiesis in Down syndrome. Blood. 2013;121(16):3181-4
Blood. 2013;121(16):3181-4
53. Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, et al. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010;24(15):1659-72
54. Li Z, Godinho FJ, Klusmann JH, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37(6):613-9
55. Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS, et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112(12):4503-6
56. Tunstall-Pedoe O, Roy A, Karadimitris A, de la Fuente J, Fisk NM, Bennett P, et al. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112(12):4507-11
Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112(12):4507-11
57. Roy A, Cowan G, Mead AJ, Filippi S, Bohn G, Chaidos A, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci USA. 2012;109(43):17579-84
58. Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, et al. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012; 109(43):17573-8
59. Izraeli S. Trisomy 21 tilts the balance. Blood. 2008;112(12):4361-2
60. MacLean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, et al. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic induced pluripotent cells. Proc Natl Acad Sci USA. 2012;109(43):17567-72
Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic induced pluripotent cells. Proc Natl Acad Sci USA. 2012;109(43):17567-72
61. Kusters MA, Verstegen RH, Gemen EF, de Vries E. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol. 2009;156(2):189-93
62. Whitlock JA, Sather HN, Gaynon P, Robison LL, Wells RJ, Trigg M, et al. Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children’s Cancer Group study. Blood. 2005; 106(13):4043-9
63. Zhang CC, Lodish HF. Insulin-like growth factor 2 expressed in a novel fetal liver population is a growth factor for hematopoietic stem cells. Blood. 2004;103(7):2513-21
64. Garrett RW, Emerson SG. The role of parathyroid hormone and insulin-like growth factors in hematopoietic niches: physiology and pharmacology. Mol Cell Endocrinol. 2006;288(1-2):6-10
Garrett RW, Emerson SG. The role of parathyroid hormone and insulin-like growth factors in hematopoietic niches: physiology and pharmacology. Mol Cell Endocrinol. 2006;288(1-2):6-10
65. Chou S, Lodish HF. Fetal liver hepatic progenitors are supportive stromal cells for hematopoietic stem cells. Proc Natl Acad Sci USA. 2010;107(17):7799-804
66. Starc TJ. Erythrocyte macrocytosis in infants and children with Down syndrome. J Pediatr. 1992;121(4):578-81
67. Kivivuori SM, Rajantie J, Siimes MA. Peripheral blood cell counts in infants with Down’s syndrome. Clin Genet. 1996;49(1):15-9
68. Henry E, Walker D, Wiedmeier SE, Christensen RD. Hematological abnormalities during the first week of life among neonates with Down syndrome: data from a multihospital healthcare system. Am J Med Genet A. 2007;143A(1):42-50
Am J Med Genet A. 2007;143A(1):42-50
69. Douglas SD. Down syndrome: immunologic and epidemiologic associations- enigmas remain. J Pediatr. 2005;147(6):723-5
70. de Hingh YC, van der Vossen PW, Gemen EF, Mulder AB, Hop WC, Brus F, et al. Intrinsic abnormalities of lymphocyte counts in children with down syndrome. J Pediatr. 2005;147(6):744-7
71. Verstegen RH, Kusters MA, Gemen EF, De Vries E. Down syndrome B-lymphocyte subpopulations: intrinsic defect or decreased T-lymphocyte help. Pediatr Res. 2010;67(5):563-9
72. Roizen NJ, Amarose AP. Hematologic abnormalities in children with Down syndrome. Am J Med Genet. 1993;46(5):510-2
73. David O, Fiorucci GC, Tosi MT, Altare F, Valori A, Saracco P, et al. Hematological studies in children with Down syndrome. Pediatr Hematol Oncol. 1996; 13(3):271-5
David O, Fiorucci GC, Tosi MT, Altare F, Valori A, Saracco P, et al. Hematological studies in children with Down syndrome. Pediatr Hematol Oncol. 1996; 13(3):271-5
74. Lin SJ, Wang JY, Klickstein LB, Chuang KP, Chen JY, Lee JF, et al. Lack of age- associated LFA-1 up-regulation and impaired ICAM-1 binding in lymphocytes from patients with Down syndrome. Clin Exp Immunol. 2001;126(1):54-63
75. Garrison MM, Jeffries H, Christakis DA. () Risk of death for children with Down syndrome and sepsis. J Pediatr. 2005;147(6):748-52
76. Gillespie KM, Dix RJ, Williams AJ, Newton R, Robinson ZF, Bingley PJ, et al. Islet autoimmunity in children with Down’s syndrome. Diabetes. 2006;55(11): 3185-8
77. Prasher VP. Screening of medical problems in adults with Down syndrome. Downs Syndr Res Pract. 1994;2:59-66
Prasher VP. Screening of medical problems in adults with Down syndrome. Downs Syndr Res Pract. 1994;2:59-66
78. Kirsammer G, Jilani S, Liu H, Davis E, Gurbuxani S, Le Beau MM, et al. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome. Blood. 2008;111(2):767-75
79. Carmichael CL, Majewski IJ, Alexander WS, Metcalf D, Hilton DJ, Hewitt CA, et al. Hematopoietic defects in the Ts1Cje mouse model of Down syndrome. Blood. 2009;113(9):1929-37
80. Alford KA, Slender A, Vanes L, Li Z, Fisher EM, Nizetic D, et al. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010; 115(14):2928-37
81. McLean S, McHale C, Enright H. Hematological abnormalities in adult patients with Down’s syndrome. Ir J Med Sci. 2009;178(1):35-8
Ir J Med Sci. 2009;178(1):35-8
82. Karlsson B, Gustafsson J, Hedov G, Ivarsson SA, Annerén G. Thyroid dysfunction in Down’s syndrome: relation to age and thyroid autoimmunity. Arch Dis Child. 1998;79(3):242-5
83. Loh RK, Harth SC, Thong YH, Ferrante A. Immunoglobulin G subclass deficiency and predisposition to infection in Down’s syndrome. Pediatr Infect Dis J. 1990;9(8):547-51
84. Murphy M, Epstein LB. Down syndrome (trisomy 21) thymuses have a decreased proportion of cells expressing high levels of TCR alpha, beta and CD3. A possible mechanism for diminished T cell function in Down syndrome. Clin Immunol Immunopathol. 1990;55(3):453-67
85. Izraeli S. Chromosome copy number and leukemia-lessons from Down’s syndrome. Hematology. 2005:10(Suppl. 1):164-6
Hematology. 2005:10(Suppl. 1):164-6
86. Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature. 2014;508(7496):345-50
87. Malinge S, Chlon T, Doré LC, Ketterling RP, Tallman MS, Paietta E, et al. Development of acute megakaryoblastic leukemia in Down syndrome is associated with sequential epigenetic changes. Blood. 2013;122(14):e33-43
88. Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone h4 Lys27 trimethylation. Nat Genet. 2014;46(6):618-23
89. Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA. 2009;106(29):12031-6
The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA. 2009;106(29):12031-6
90. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megaka- ryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012; 122(3):948-62
91. Ng AP, Hyland CD, Metcalf D, Carmichael CL, Loughran SJ, Di Rago L, et al. Trisomy of Erg is required for myeloproliferation in a mouse model of Down syndrome. Blood. 2010;115(19):3966-9
92. Birger Y, Goldber L, Chlon TM, Goldenson B, Muler I, Schiby G, et al. Perturbation of fetal hematopoiesis in a mouse model of Down syndrome’s transient myeloproliferative disorder. Blood. 2013;122(6):988-98
Blood. 2013;122(6):988-98
93. Bourquin JP, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci USA. 2006;103(9): 3339-44
94. Gribble SM, Wiseman FK, Clayton S, Prigmore E, Langley E, Yang F, et al. Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of Down syndrome. PLoS One. 2013;8(4):e60482
95. Dauphinot L, Lyle R, Rivals I, Dang MT, Moldrich RX, Golfier G, et al. The cerebellar transcriptome during postnatal development of the Ts1Cje mouse, a segmental trisomy model for Down syndrome. Hum Mol Genet. 2005;14(3):373-84
96. Aït-Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, Golfier G, et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet. 2007;81(3):475-91
Aït-Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, Golfier G, et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet. 2007;81(3):475-91
97. Conti A, Fabbrini F, D’Agostino P, Negri R, Greco D, Genesio R, et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genomics. 2007;8:268
98. Hertzberg L, Betts DR, Raimondi SC, Schäfer BW, Notterman DA, Domany E, et al. Prediction of chromosomal aneuploidy from gene expression data. Genes Chromosomes Cancer. 2007;46(1):75-86
99. Prandini P, Deutsch S, Lyle R, Gagnebin M, Delucinge Vivier C, Delorenzi M, et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet. 2007;81(2):252-63
Am J Hum Genet. 2007;81(2):252-63
100. Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007; 90(6):647-60
101. Gefen N, Binder V, Zaliova M, Linka Y, Morrow M, Novosel A, et al. Hsa-mir-125b-2 is highly expressed in childhood ETV6/RUNX1 (TEL/AML1) leukemias and confers survival advantage to growth inhibitory signals independent of p53. Leukemia. 2010;24(1):89-96
102. Emmrich S, Rasche M, Schöning J, Reimer C, Keihani S, Maroz A, et al. miR-99a/100~125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGF and Wnt signaling. Genes Dev. 2014;28(8):858-74
103. Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM, et al. Translating dosage compensation to trisomy 21. Nature. 2014;500(7462):296-300
Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM, et al. Translating dosage compensation to trisomy 21. Nature. 2014;500(7462):296-300
104. Elagib KE, Racke FK, Mogass M, Khetawat R, Delehanty LL, Goldfarb AN. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood. 2003;101(11):4333-41
105. Rainis L, Toki T, Pimanda JE, Rosenthal E, Machol K, Strehl S, et al. The proto- oncogene ERG in megakaryoblastic leukemias. Cancer Res. 2005;65(17):7596-602
106. Xu G, Kanezaki R, Toki T, Watanabe S, Takahashi Y, Terui K, et al. Physical association of the patient-specific GATA1 mutants with RUNX1 in acute mega- karyoblastic leukemia accompanying Down syndrome. Leukemia. 2006; 20(6):1002-8
107. Yu S, Cui K, Jothi R, Zhao DM, Jing X, Zhao K, et al. GABP controls a critical transcription regulatory module that is essential for maintenace and differentiation of hematopietic stem/progenitor cells. Blood. 2011;117(7):2166-78
Yu S, Cui K, Jothi R, Zhao DM, Jing X, Zhao K, et al. GABP controls a critical transcription regulatory module that is essential for maintenace and differentiation of hematopietic stem/progenitor cells. Blood. 2011;117(7):2166-78
108. Garzon R, Pichiorri RF, Palumbo T, Iuliano R, Cimmino A, Aqeilan R, et al. MicroRNA fingerprints during human megakaryocytopoiesis. Proc Natl Acad Sci USA. 2006;103(13):5078-83
109. Klusmann JH, Li Z, Böhmer K, Maroz A, Koch ML, Emmrich S, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Gen Dev. 2010;24(5):478-90
110. Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, et al. RUNX1 mutations are frequent in de novo AML with non-complex karyotype and confer an unfavorable prognosis. Blood. 2011;117(8):2348-57
Blood. 2011;117(8):2348-57
ДАУНА СИНДРОМ | Энциклопедия Кругосвет
Содержание статьи
ДАУНА СИНДРОМ, врожденное нарушение развития, проявляющееся умственной отсталостью, нарушением роста костей и другими физическими аномалиями. Это одна из наиболее распространенных форм умственной отсталости; ею страдает примерно 10% больных, поступающих в психиатрические лечебницы. Для больных с синдромом Дауна характерно сохранение физических черт, свойственных ранней стадии развития плода, в том числе узких раскосых глаз, придающих больным внешнее сходство с людьми монголоидной расы, что дало основание Л.Дауну назвать в 1866 данное заболевание «монголизмом» и предложить ошибочную теорию расовой регрессии, или эволюционного отката. На самом деле синдром Дауна не связан с расовыми особенностями и встречается у представителей всех рас. Синдром удалось экспериментально воспроизвести у крыс путем рентгеновского облучения эмбриона на 12–13-й день беременности.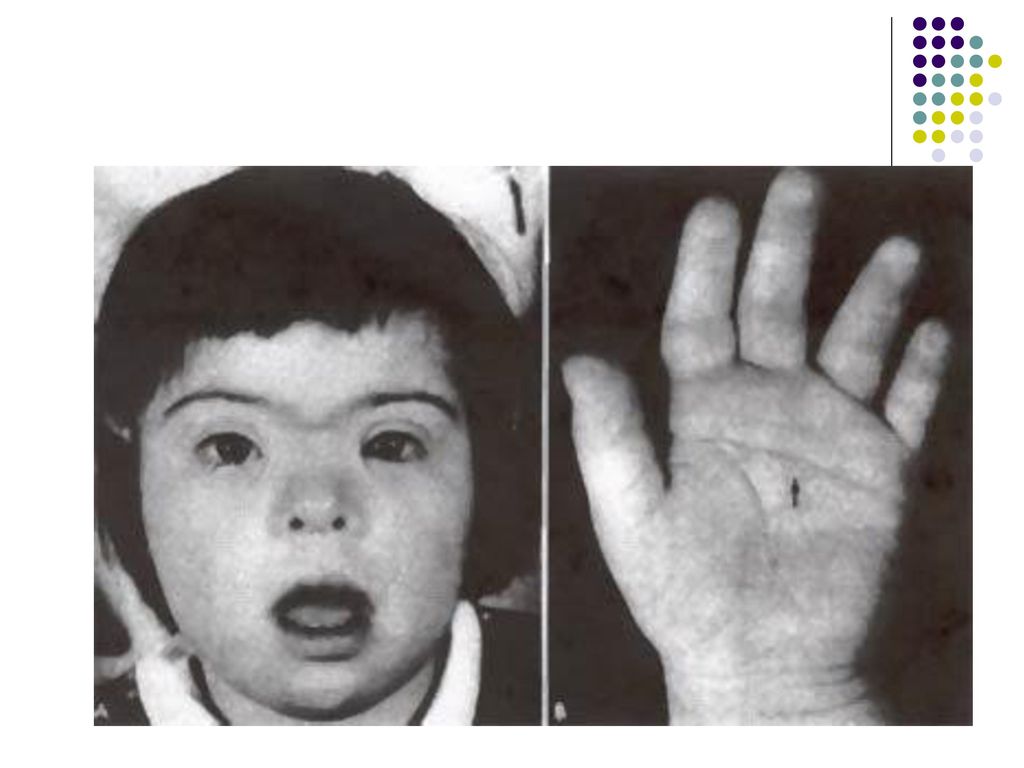
Характеристика.
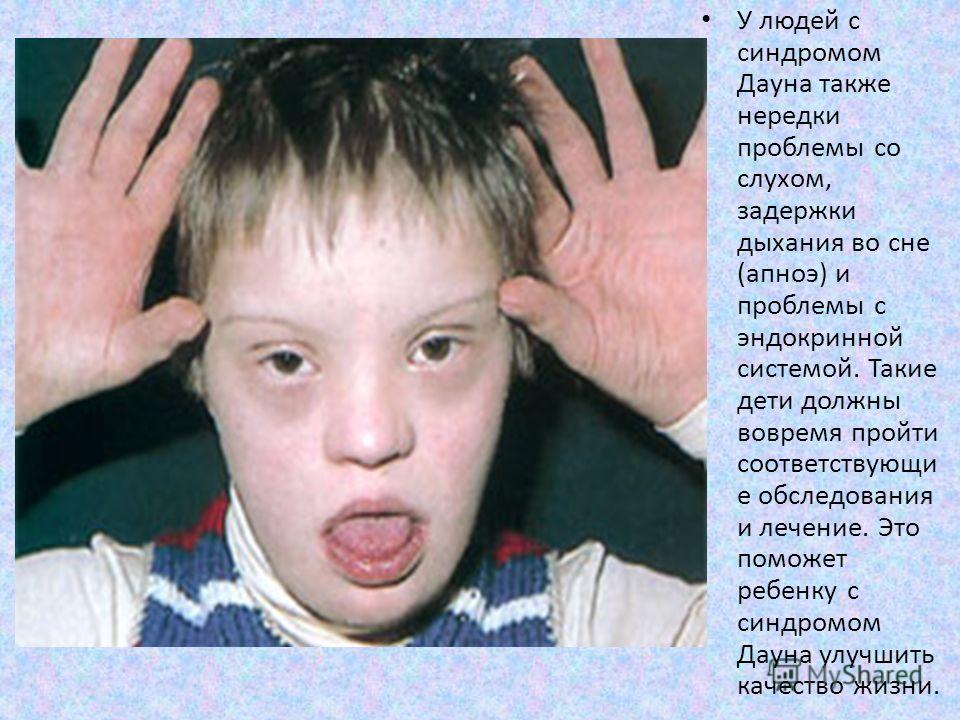
Помимо уже упоминавшихся особенностей строения глаз у больных с синдромом Дауна выявляются и другие характерные признаки: маленькая округлая голова, гладкая влажная отечная кожа, сухие истонченные волосы, маленькие округлые уши, маленький нос, толстые губы, поперечные бороздки на языке, который зачастую высунут наружу, так как не помещается в полости рта. Пальцы короткие и толстые, мизинец сравнительно мал и обычно загнут вовнутрь. Расстояние между первым и вторым пальцами на кистях и стопах увеличено. Конечности короткие, рост, как правило, значительно ниже нормы. Половые признаки развиты слабо, и, вероятно, в большинстве случаев способность к репродукции отсутствует.
Интеллект больных обычно снижен до уровня умеренной умственной отсталости. Коэффициент интеллектуального развития (IQ) колеблется между 20 и 49, хотя в отдельных случаях может быть выше или ниже этих пределов. Даже у взрослых больных умственное развитие не превышает уровень нормального семилетнего ребенка. В руководствах традиционно описываются такие черты больных с синдромом Дауна, как покорность, позволяющая им хорошо приспосабливаться к больничной жизни, ласковость, сочетающиеся с упрямством, отсутствием гибкости, склонность к подражательству, а также чувство ритма и любовь к танцам. Однако систематические исследования, проведенные в Англии и США, не подтверждают этот образ.
В руководствах традиционно описываются такие черты больных с синдромом Дауна, как покорность, позволяющая им хорошо приспосабливаться к больничной жизни, ласковость, сочетающиеся с упрямством, отсутствием гибкости, склонность к подражательству, а также чувство ритма и любовь к танцам. Однако систематические исследования, проведенные в Англии и США, не подтверждают этот образ.
Причины.
В качестве возможных причин синдрома Дауна рассматривались многие факторы, но в настоящее время твердо установлено, что в основе его лежит аномалия хромосом: лица, страдающие этим расстройством, имеют, как правило, 47 хромосом вместо нормальных 46. Дополнительная хромосома является результатом нарушенного созревания половых клеток. В норме при делении незрелых половых клеток парные хромосомы расходятся, и каждая зрелая половая клетка получает 23 хромосомы. Во время оплодотворения, т.е. слияния материнской и отцовской клетки, нормальный набор хромосом восстанавливается (см. также ЭМБРИОЛОГИЯ). В основе синдрома Дауна лежит нерасхождение одной из хромосомных пар, обозначаемой как 21-я. В результате у ребенка появляется лишняя (третья) 21-я хромосома. Это состояние называется трисомией по 21-й паре хромосом. Хотя в подавляющем большинстве случаев при синдроме Дауна обнаруживается именно эта трисомия, крайне редко встречаются и другие хромосомные аномалии.
Генетические исследования на плодовых мушках (дрозофилах) показали, что важнейшим фактором, определяющим нерасхождение хромосом при созревании яйцеклетки, является возраст матери. В отношении синдрома Дауна уже давно было известно, что вероятность рождения больного ребенка растет с увеличением возраста матери, причем тем быстрее, чем она старше. Число детей с этим синдромом, появившихся у матерей после 35 лет, значительно выше, чем у более молодых. По этой причине врачи часто советуют будущим матерям, чей возраст превышает 35 лет, прибегнуть к амниоцентезу, т.е. процедуре получения образца околоплодных вод для анализа хромосомного состава клеток. Это дает возможность прервать беременность, угрожающую рождением больного ребенка.
Это дает возможность прервать беременность, угрожающую рождением больного ребенка.
Установлено, что если синдромом Дауна страдает один из однояйцовых близнецов, то неизбежно болен и другой, а у разнояйцовых близнецов, как и вообще у братьев и сестер, вероятность такого совпадения значительного ниже. Данный факт дополнительно свидетельствует в пользу хромосомного происхождения болезни. Однако синдром Дауна нельзя считать наследственным заболеванием, так как при нем не происходит передачи дефектного гена из поколения в поколение, а расстройство возникает на уровне репродуктивного процесса.
Лечение.
Предпринимались попытки лечить детей с синдромом Дауна гормонами щитовидной железы и гипофиза, однако эти методы находятся пока на стадии разработки. Как и другие умственно отсталые дети их уровня, больные с синдромом Дауна поддаются обучению бытовым навыкам, координации движений, речи и другим простым функциям, необходимым в повседневной жизни.
См. также УМСТВЕННАЯ ОТСТАЛОСТЬ.
Проверь себя!
Ответь на вопросы викторины «Фобии и страхи»
Чего больше всего боятся трискаидекафобы?
COVID-19 у пациентов с синдромом Дауна
Оригинал: Acta Paediatrica
Автор: Michele Callea et al.
Опубликовано: 13 июня 2020, Acta Paediatrica
Перевод: Олеся Кузнецова, Фонд профилактики рака
Вспышка тяжелой пневмонии, вызванной новым вирусом SARS‑CoV‑2, создала чрезвычайную ситуацию во всем мире. За время пандемии опубликовано более 10 000 статей о COVID‑19, но ни в одной из них не рассматривается влияние COVID‑19 на пациентов с синдромом Дауна.
COVID‑19 проявляется как тяжелый острый респираторный синдром, а синдром Дауна — наиболее распространенное хромосомное заболевание, с высоким риском респираторных инфекций и их осложнений [1]. Это важно учитывать во время пандемии. Частота синдрома Дауна среди новорожденных составляет 1:700. Продолжительность жизни таких пациентов увеличивается, и многие достигают взрослого возраста [1].
Продолжительность жизни таких пациентов увеличивается, и многие достигают взрослого возраста [1].
Считается, что дети менее подвержены COVID‑19. Однако дети с синдромом Дауна — это уязвимая группа, они в большей степени подвержены респираторным инфекциям. Кроме этого, у детей с синдромом Дауна чаще имеются сопутствующие заболевания: иммунодефициты, заболевания сердца (в том числе патологии, требующие протезирования клапанов), ожирение и диабет. Известно, что перечисленные заболевания ухудшают исходы COVID‑19 [3].
Лечение COVID‑19 остается эмпирическим. Для профилактики и коррекции осложнений используют различные комбинации препаратов: противовирусные средства (ремдесивир), противовоспалительные препараты (хлорохин, азитромицин) и антикоагулянты [4].
Пациентам с синдромом Дауна рекомендуют соблюдать стандартные меры профилактики: социальное дистанцирование, использование защитных масок и перчаток, частое мытье рук, дезинфекция рук и поверхностей.
Семьи, воспитывающие детей с синдромом Дауна, во время пандемии испытывают особые трудности. Примерно в 40 % таких семей один из родителей (или другой член семьи) не работал на момент начала пандемии. Другие члены семьи продолжают работать и, следовательно, увеличивают риск инфицирования. Ритм жизни в семьях нарушается: резко сокращаются социальные контакты, а дети с синдромом Дауна лишаются общения (обычно они с удовольствием ходят в школу и любят общаться со старшими родственниками).
Примерно в 40 % таких семей один из родителей (или другой член семьи) не работал на момент начала пандемии. Другие члены семьи продолжают работать и, следовательно, увеличивают риск инфицирования. Ритм жизни в семьях нарушается: резко сокращаются социальные контакты, а дети с синдромом Дауна лишаются общения (обычно они с удовольствием ходят в школу и любят общаться со старшими родственниками).
Центр поддержки детей с синдромом Дауна больницы Bambino Gesù предлагает ряд рекомендаций:
— Обеспечить детям с синдромом Дауна ранний доступ к диагностическим тестам и противовирусной терапии (это особенно важно для пациентов с нарушениями иммунитета, врожденными пороками сердца, ожирением).
— Для ОТ-ПЦР лучше использовать слюну (для взятия пробы пациент сплевывает в стерильную емкость), а не мазок из носо- и ротоглотки. Тестирование слюны безопасно, неинвазивно и обладает такой же диагностической ценностью, как и мазок [5].
— Объяснить семьям, что о любых подозрительных симптомах нужно сообщить лечащему врачу и/или врачу центра (см. схему на рис. 1). Необходимо соблюдать особую осторожность, чтобы избежать бытовых травм и, соответственно, обращений в травмпункты или отделения неотложной помощи, а также возможных госпитализаций. Госпитализация в отделение интенсивной терапии сопровождается особенно высоким риском.
схему на рис. 1). Необходимо соблюдать особую осторожность, чтобы избежать бытовых травм и, соответственно, обращений в травмпункты или отделения неотложной помощи, а также возможных госпитализаций. Госпитализация в отделение интенсивной терапии сопровождается особенно высоким риском.
— Центр готов предоставить семьям, воспитывающим детей с синдромом Дауна, телемедицинскую консультацию по любому вопросу.
Список литературы
- Valentini D, Di Camillo C, Mirante N, et al. Effects of Pidotimod on recurrent respiratoryinfections in children with Down syndrome: a retrospective Italian study. Ital J Pediatr.2020; 46: 31.
- Brodin P. Why is COVID-19 so mild in children? Acta Paediatr. 2020; 109: 1082-3.
- Siordia JA Jr. Epidemiology and clinical features of COVID-19: A review of currentliterature. J Clin Virol. 2020; 127: 104357.
- Some drugs for COVID-19. Med Lett Drugs Ther. 2020; 62: 49‐50.
- Sri Santosh T, Parmar R, Anand H, Srikanth K, Saritha M.
 A review of salivarydiagnostics and its potential implication in detection of COVID-19. Cureus. 2020; 12:e7708.
A review of salivarydiagnostics and its potential implication in detection of COVID-19. Cureus. 2020; 12:e7708.
 A review of salivarydiagnostics and its potential implication in detection of COVID-19. Cureus. 2020; 12:e7708.
A review of salivarydiagnostics and its potential implication in detection of COVID-19. Cureus. 2020; 12:e7708.закрыть меню
Синдром Дауна в практике гематолога
SOVREMENNAYA PEDIATRIYA.2017.6(86):130-146; doi 10.15574/SP.2017.86.130
Дорош О. И., Трояновская О. О., Очеретная О. М., Кицера Н. И., Иваненко А. Л., Середич Л. П., Мых А. М., Цымбалюк-Волошин И. П., Бескоровайная Г. М., Мельничук Л. В., Тысячна Л. М., Микула М. И., Бидюк В. М., Степанюк А. И., Козлова О. И., Дубей Л. Я., Полищук Р. С., Скоропад Л. Л., Воробель О. И., Савчак И. Я.
КЗ ЛОР «Западноукраинский специализированный детский медицинский центр», г. Львов, Украина
Львовский национальный медицинский университет имени Данила Галицкого, Украина
Николаевская областная детская больница, Украина
Областная детская клиническая больница, г. Ивано—Франковск, Украина
Львовская областная клиническая больница, Украина
ГУ «Институт наследственной патологии НАМН Украины», г. Львов
Львов
Представлен анализ клинико-лабораторных проявлений, особенностей кроветворения у 26 детей с синдромом Дауна (СД). Установлено, что детям с СД присущи различные врожденные пороки развития с высокой частотой врожденных пороков сердца — 69,2%, с преобладанием полной атриовентрикулярной коммуникации (ПАВК) и дефекта межжелудочковой перегородки. Выяснено, что среди гематологических расстройств при трисомии 21-й хромосомы 30,8% составляет железодефицитная анемия; транзиторный анормальный миелопоэз (ТАМ) встречается у 19,2% больных, иммунная тромбоцитопеническая пурпура — в 15,4% случаев. У трех из пяти детей с ТАМ зарегистрированы клональные заболевания: миелодиспластический синдром, идиопатический миелофиброз и ганглионейробластома. Это дает основания считать, что ТАМ является отдельной нозологической единицей, переходным миелопролиферативным расстройством, которое регрессирует самостоятельно у двух из пяти больных бесследно, хотя и может быть предвестником других серьезных заболеваний. Детям с СД присущи злокачественные заболевания крови: острая миелоидная лейкемия (ОМЛ) верифицирована у 15,4% пациентов, острый лимфобластный лейкоз — у 7,7% детей. Опухоли негематологического происхождения и неходжкинская лимфома у детей с трисомией 21-й хромосомы встречаются с небольшой частотой (3,8%). Установлено, что для детей с СД, больных ОМЛ, протокольная химиотерапия является високоэффективным методом лечения. У больных гемобластозами В-линейного происхождения при проведении химиотерапии с применением метотрексата независимо от дозы (0,5 г/м2 или 2 г/м2) встречаются тяжелые токсические поражения кожи, слизистых оболочек, висцеральных органов. Показатель общего кумулятивного выживания (риск смерти) у детей с СД составил 0,68.
Детям с СД присущи злокачественные заболевания крови: острая миелоидная лейкемия (ОМЛ) верифицирована у 15,4% пациентов, острый лимфобластный лейкоз — у 7,7% детей. Опухоли негематологического происхождения и неходжкинская лимфома у детей с трисомией 21-й хромосомы встречаются с небольшой частотой (3,8%). Установлено, что для детей с СД, больных ОМЛ, протокольная химиотерапия является високоэффективным методом лечения. У больных гемобластозами В-линейного происхождения при проведении химиотерапии с применением метотрексата независимо от дозы (0,5 г/м2 или 2 г/м2) встречаются тяжелые токсические поражения кожи, слизистых оболочек, висцеральных органов. Показатель общего кумулятивного выживания (риск смерти) у детей с СД составил 0,68.
Ключевые слова: дети, синдром Дауна, транзиторный анормальный миелопоэз, анемия, острый лимфобластный лейкоз, острый миелоидный лейкоз, врожденные пороки развития.
Литература
1. Динаміка частоти синдрому Дауна у Львівській області за 1985—2001 роки / О. З. Гнатейко, Д.В. Заставна, Н.І. Кіцера [та ін.] // Перинатологія та педіатрія. — 2003. — №2. — С. 50—53.
З. Гнатейко, Д.В. Заставна, Н.І. Кіцера [та ін.] // Перинатологія та педіатрія. — 2003. — №2. — С. 50—53.
2. Населення Львівської області [Електронний ресурс] // Вікіпедія. — Режим доступу : https://uk.wikipedia.org/wiki/ (дата звернення: 10.05.17). — Назва з екрану.
3. Поширеність хромосомної патології серед дитячої популяції Чернівецької області / Т.М. Бойчук, Т.В. Сорокман, І.В. Ластівка, М.О. Ризничук // Буковинський медичний вісник. — 2011. — Т.15, №1(57). — С. 24—29.
4. Синдром Дауна (синдром трисомії 21) [Електронний ресурс] / О.О. Кисельова, Л.С. Євтушок // Medicus Amicus. — 2006. — №2. — C. 15. — Режим доступу : http://www.ibis-birthdefects.org/start/ukrainian/udown.htm (дата звернення: 10.05.17).
5. Depince-Berger A, Cremilieux C, Rinaudo-Gaujous M et al. (2016). A difficult and rare diagnosis of autoimmune enteropathy in a patient affected by Down syndrome. J Clin Immunol. 36(5): 423-428. https://doi.org/10.1007/s10875-016-0280-7; PMid:27072857
6. Satgé D, Sasco AJ, Carlsen NL et al. (1998). A lack of neuroblastoma in Down syndrome: a study from 11 European countries. Cancer Res. 58(3): 448-452. PMid:9458088
Satgé D, Sasco AJ, Carlsen NL et al. (1998). A lack of neuroblastoma in Down syndrome: a study from 11 European countries. Cancer Res. 58(3): 448-452. PMid:9458088
7. Satgé D, Sommelet D, Geneix A et al. (1998). A tumor profile in Down syndrome. Am J Med Genet. 78(3): 207-216. https://doi.org/10.1002/(SICI)1096-8628(19980707)78:3<207::AID-AJMG1>3.0.CO;2-M
8. Trudy D Buitenkamp, Shai Izraeli, Martin Zimmermann et al. (2014). Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 123: 70-77. https://doi.org/10.1182/blood-2013-06-509463; PMid:24222333 PMCid:PMC3879907
9. Kojima S, Sako M, Kato K et al. (2000). An effective chemotherapeutic regimen for acute myeloid leukemia and myelodysplastic syndrome in children with Down’s syndrome. Leukemia. 14(5): 786-791. https://doi.org/10.1038/sj.leu.2401754; PMid:10803507
10. Steiner B, Masood R, Rufibach K et al. (2015). An unexpected finding: younger fathers have a higher risk for offspring with chromosomal aneuploidies. Eur J Hum Genet. 23(4): 466-472. https://doi.org/10.1038/ejhg.2014.122; PMid:25005732 PMCid:PMC4666566
11. Tenenbaum A, Malkiel S, Wexler ID et al. (2011). Anemia in children with Down syndrome. Int J Pediatr. 2011: ID 813541.
12. Yancey CL, Zmijewski C, Athreya BH, Doughty RA. (1984). Arthropathy of Down’s syndrome. Arthritis Rheum. 27(8): 929-934. https://doi.org/10.1002/art.1780270813; PMid:6235815
13. Satgé D, Sasco AJ, Vekemans MJ et al. (2006). Aspects of digestive tract tumors in Down syndrome: a literature review. Dig Dis Sci. 51(11): 2053-2061. https://doi.org/10.1007/s10620-006-9131-3; PMid:17009117
14. Szaflarska-Popławska A, Soroczyńska-Wrzyszcz A, Barg E et al. (2016). Assessment of coeliac disease prevalence in patients with Down syndrome in Poland – a multicentre study. Prz Gastroenterol. 11(1): 41-46. https://doi.org/10.5114/pg.2016.57794; PMid:27110310 PMCid:PMC4814541
15. Sotonica M, Mackic-Djurovic M, Hasic S et al. (2016). Association of parental age and the type of Down syndrome on the territory of Bosnia and Herzegovina. Med Arch. 70(2): 88-91. https://doi.org/10.5455/medarh.2016.70.88-91; PMid:27147778 PMCid:PMC4851533
16. Bain B. (1991). Down’s syndrome-transient abnormal myelopoiesis and acute leukaemia. Leuk Lymphoma. 3(5-6): 309-317. https://doi.org/10.3109/10428199109070274; PMid:27467421
17. Bruwier A, Chantrain CF. (2012). Hematological disorders and leukemia in children with Down syndrome. Eur J Pediatr. 171(9): 1301-1307. https://doi.org/10.1007/s00431-011-1624-1; PMid:22113227
18. Butler DR, Chilvers CR, Cane RJ. (2007). The implications and management of acute odontogenic infection in association with Down and Eisenmenger syndromes and schizophrenia in a rural setting. Aust Dent J. 52(1): 61-66. https://doi.org/10.1111/j.1834-7819.2007.tb00467.x; PMid:17500166
19. Choi JK. (2008). Hematopoietic disorders in Down syndrome. Int J Clin Exp Pathol. 1(5): 387-395. PMid:18787621 PMCid:PMC2480572
20. Chung EM, Sung EC, Sakurai KL. (2004). Dental management of the Down and Eisenmenger syndrome patient. J Contemp Dent Pract. 5(2): 70-80. PMid:15150635
21. Jackson GL, Sendelbach DM, Rambally B et al. (2012). Circulating blasts and associated hematologic disorders in neonates with Down syndrome. Am J Perinatol. 29(4): 259-266. https://doi.org/10.1055/s-0031-1285103; PMid:21809264
22. Ceppi F, Stephens D, den Hollander BS et al. (2016). Clinical presentation and risk factors of serious infections in children with Down syndrome treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 63(11): 1949-1953. https://doi.org/10.1002/pbc.26127; PMid:27399585
23. Saida S, Watanabe K, Sato-Otsubo A et al. (2013). Clonal selection in xenografted TAM recapitulates the evolutionary process of myeloid leukemia in Down syndrome. Blood. 121: 4377-4387. https://doi.org/10.1182/blood-2012-12-474387; PMid:23482930
24. Pavlović M, Radlović N, Leković Z et al. (2010). Coeliac disease as the cause of resistant sideropenic anaemia in children with Down’s syndrome: case report. Srp Arh Celok Lek. 138(1-2): 91-94. https://doi.org/10.2298/SARh2002091P; PMid:20422917
25. Coppedè F. (2016). Risk factors for Down syndrome. Arch Toxicol. 90(12): 2917-2929. https://doi.org/10.1007/s00204-016-1843-3; PMid:27600794
26. Sharr C, Lavigne J, Elsharkawi IM et al. 2016. Detecting celiac disease in patients with Down syndrome. Am J Med Genet A. 170(12): 3098-3105. https://doi.org/10.1002/ajmg.a.37879; PMid:27605215
27. Chen J, Yousif F, Beck T et al. (2014). Differences of somatic mutations and gene expression in blasts of transient leukemia and acute myeloid leukemia of Down syndrome. Blood. 124: 2364.
28. Duffels MG, Vis JC, van Loon RL et al. (2009). Down patients with Eisenmenger syndrome: is bosentan treatment an option? Int J Cardiol. 134(3): 378-383. https://doi.org/10.1016/j.ijcard.2008.02.025; PMid:18579234
29. Asim A, Kumar A, Muthuswamy S et al. (2015). Down syndrome: an insight of the disease. J Biomed Sci. 22(1): 41-49. https://doi.org/10.1186/s12929-015-0138-y; PMid:26062604 PMCid:PMC4464633
30. Concolino D, Pasquzzi A, Capalbo G. et al. (2006). Early detection of podiatric anomalies in children with Down syndrome. Acta Paediatr. 95(1): 17-20. https://doi.org/10.1111/j.1651-2227.2006.tb02174.x; https://doi.org/10.1080/08035250500325108; PMid:16373291
31. Körten MA, Helm PC, Abdul-Khaliq H et al. (2016). Defects Investigators Eisenmenger syndrome and long-term survival in patients with Down syndrome and congenital heart disease. Competence Network for Congenital Heart. Heart. 102(19): 1552-1557. https://doi.org/10.1136/heartjnl-2016-309437; PMid:27325590
32. Kuitert PC, Abbink FC, Broers CJ et al. (2014). Epstein-Barr virus infection with severe consequences. EBV, haemophagocytic lymphohistiocytosis and Hodgkin lymphoma with Down syndrome. Ned Tijdschr Geneeskd. 158: A7608. PMid:24988166
33. Evans DI. (1975). Acute myelofibrosis in children with Down’s syndrome. Arch Dis Child. 50(6): 458-462. https://doi.org/10.1136/adc.50.6.458; PMid:125073 PMCid:PMC1544537
34. Hasle H, Clemmensen IH, Mikkelsen M. (2000). Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 355(9199): 165-169. https://doi.org/10.1016/S0140-6736(99)05264-2
35. Henry E, Walker D, Wiedmeier SE, Christensen RD. (2007). Hematological abnormalities during the first week of life among neonates with Down syndrome: data from a multihospital healthcare system. Am J Med Genet A. 143A(1): 42-50. https://doi.org/10.1002/ajmg.a.31442; PMid:17163522
36. Awasthi A, Das R, Varma N et al. (2005). Hematological disorders in Down syndrome: ten-year experience at a Tertiary Care Centre in North India. Pediatr Hematol Oncol. 22(6): 507-512. https://doi.org/10.1080/08880010591002350; PMid:16169817
37. Baraona F, Gurvitz M, Landzberg MJ, Opotowsky AR. (2013). Hospitalizations and mortality in the United States for adults with Down syndrome and congenital heart disease. Am J Cardiol. 111(7): 1046-1051. https://doi.org/10.1016/j.amjcard.2012.12.025; PMid:23332593
38. Aversa T, Valenzise M, Corrias A et al. (2016). In children with autoimmune thyroid diseases the association with Down syndrome can modify the clustering of extra-thyroidal autoimmune disorders. J Pediatr Endocrinol Metab. 29(9): 1041-1046. https://doi.org/10.1515/jpem-2016-0073; PMid:27442363
39. Hassler A, Bochennek K, Gilfert J et al. (2016). Infectious complications in children with acute myeloid leukemia and Down syndrome: analysis of the prospective multicenter trial AML-BFM 2004. Pediatr Blood Cancer. 63(6): 1070-1074. https://doi.org/10.1002/pbc.25917; PMid:26814618
40. Irving CA, Chaudhari MP. (2012). Cardiovascular abnormalities in Down’s syndrome: spectrum, management and survival over 22 years. Arch Dis Child. 97: 326-330. https://doi.org/10.1136/adc.2010.210534; PMid:21835834
41. Kawatu D, LeLeiko NS. (2006). Screening for celiac disease in asymptomatic children with Down syndrome: cost-effectiveness of preventing lymphoma. Pediatrics. 118(2): 816-817. https://doi.org/10.1542/peds.2006-1194; PMid:16882844
42. Khan I, Malinge S, Crispino JD. (2011). Myeloid leukemia in Down syndrome. Crit Rev Oncog. 16(1-2): 25-36. https://doi.org/10.1615/CritRevOncog.v16.i1-2.40; PMid:22150305 PMCid:PMC3243928
43. Kivivuori SM, Rajantie J, Siimes MA. (1996). Peripheral blood cell counts in infants with Down’s syndrome. Clin Genet. 49(1): 15-19. https://doi.org/10.1111/j.1399-0004.1996.tb04318.x; PMid:8721566
44. Duque Orozco MD, Abousamra O, Chen BP et al. (2016). Knee deformities in children with Down syndrome: a focus on knee malalignment. J Pediatr Orthop. Epub ahead of print. https://doi.org/10.1097/BPO.0000000000000814
45. Crepaz R, Romeo C, Montanaro D, De Santis S. (2013). Long-term results of treatment with bosentan in adult Eisenmenger’s syndrome patients with Down’s syndrome related to congenital heart disease. BMC Cardiovasc Disord. 13: 74. https://doi.org/10.1186/1471-2261-13-74; PMid:24047157 PMCid:PMC3848635
46. Lorenzana AN, Schorin MA. (1989). Non-Hodgkin’s lymphoma in a neonate with Down’s syndrome. Case report and literature review. Am J Pediatr Hematol Oncol. 11(2): 186-190.
47. Hasle H, Friedman JM, Olsen JH, Rasmussen SA. (2016). Low risk of solid tumors in persons with Down syndrome. Genet Med. 18(11): 1151-1157.
48. Draghi F, Bonardi M, Dellabianca C et al. (2011). Lymphoma of the scrotum in patients with Down’s syndrome: US appearance. Mini-pictorial essay. J Ultrasound. 14(4): 216-219.
49. Mendez AA, Keret D, MacEwen GD. (1988). Treatment of patellofemoral instability in Down’s syndrome. Clin Orthop Relat Res. (234): 148-158.
50. Merrick J, Ezra E, Josef B еt al. (2000). Musculoskeletal problems in Down syndrome European Paediatric Orthopaedic Society Survey: the Israeli sample. J Pediatr Orthop B. 9(3): 185-192.
51. Niceta M, Stellacci E, Gripp KW еt al. (2015). Mutations impairing GSK3-Mediated MAF phosphorylation cause cataract, deafness, intellectual disability, seizures, and a Down syndrome-like facies. Am J Hum Genet. 96(5): 816-825.
52. Satomi K, Yoshida M, Matsuoka K et al. (2014). Myelopathy mimicking subacute combined degeneration in a Down syndrome patient with methotrexate treatment for B lymphoblastic leukemia: report of an autopsy case. Neuropathology. 34(4): 414-419.
53. Corone S, Davido A, Lang T, Corone P. (1992). Outcome of patients with Eisenmenger syndrome. Apropos of 62 cases followed-up for an average of 16 years. Arch Mal Coeur Vaiss. 85(5): 521-526.
54. Inaba H, Zhou Y, Abla O et al. (2014). Pediatric acute megakaryoblastic leukemia without Down syndrome: a retrospective study by the international Berlin-Frankfurt-Munster Study Group (I-BFMSG). Blood. 124: 3670.
55. Petershofer A, Fingernagel T, Trieb K. (2015). Patellofemoral instability in trisomy 21: MPFL-Reconstruction as a single procedure. Orthopade. 44(8): 643-646.
56. Prasanna Ghimire, Guang-Yao Wu, Ling Zhu. (2011). Primary gastrointestinal lymphoma. World J Gastroenterol. 17(6): 697-707.
57. Bhat AS, Chaturvedi MK, Saini S et al. (2013). Prevalence of celiac disease in Indian children with Down syndrome and its clinical and laboratory predictors. Indian J Pediatr. 80(2): 114-117.
58. Dixon NE, Crissman BG, Smith PB еt al. (2010). Prevalence of iron deficiency in children with Down syndrome. J Pediatr. 157(6): 967-971.e1.
59. Jung JG, Kang HW, Hahn SJ et al. (2013). Primary mucosa-associated lymphoid tissue lymphoma of the esophagus, manifesting as a submucosal tumor. Korean J Gastroenterol. 62(2): 117-121.
60. Ueda K, Kawaguchi Y, Kodama M et al. (1981). Primary myelofibrosis with myeloid metaplasia and cytogenetically abnormal clones in 2 children with Down’s syndrome. Scand J Haematol. 27(3): 152-158.
61. Rabin KR, Whitlock JA. (2009). Malignancy in children with trisomy 21. Oncologist. 14(2): 164-173. https://doi.org/10.1634/theoncologist.2008-0217; PMid:19176633 PMCid:PMC2761094
62. Rabin KR, Smith J, Kozinetz CA. (2012). Myelosuppression and infectious complications in children with Down syndrome and acute lymphoblastic leukemia. Pediatr Blood Cancer. 58(4): 633-635. https://doi.org/10.1002/pbc.23371; PMid:22106003 PMCid:PMC3279631
63. Ram G, Chinen J. (2011). Infections and immunodeficiency in Down syndrome. Clin Exp Immunol. 164(1): 9-16. https://doi.org/10.1111/j.1365-2249.2011.04335.x; PMid:21352207 PMCid:PMC3074212
64. Delanoë A, Lépine J, Turcotte S et al. (2016). Role of psychosocial factors and health literacy in pregnant women’s intention to use a decision aid for Down syndrome screening: a theory-based web survey. J Med Internet Res. 18(10): e283. https://doi.org/10.2196/jmir.6362; PMid:27793792 PMCid:PMC5106559
65. Swigonski NL, Kuhlenschmidt HL, Bull MJ et al. (2006). Screening for celiac disease in asymptomatic children with Down syndrome: cost-effectiveness of preventing lymphoma. Pediatrics. 118(2): 594-602. https://doi.org/10.1542/peds.2005-2123; PMid:16882812
66. Salazar EG, Li Y, Fisher BT et al. (2016). Supportive care utilization and treatment toxicity in children with Down syndrome and acute lymphoid leukaemia at free-standing paediatric hospitals in the United States. Br J Haematol. 174(4): 591-599. https://doi.org/10.1111/bjh.14085; PMid:27161549 PMCid:PMC5684439
67. Sahin M, Tutuncu NB, Kanbay M, Guvener ND. (2006). Surgery for hyperthyroidism in Down syndrome: case report. Mt Sinai J Med. 73(5): 784-786. PMid:17008939
68. Bettuzzi C, Lampasi M, Magnani M, Donzelli O. (2009). Surgical treatment of patellar dislocation in children with Down syndrome: a 3- to 11-year follow-up study. Knee Surg Sports Traumatol Arthrosc. 17(4): 334-340. https://doi.org/10.1007/s00167-008-0652-5; PMid:18974972
69. Taub JW, Ge Y. (2005). Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer. 44(1): 33-39. https://doi.org/10.1002/pbc.20092; PMid:15390307
70. De Haan M, de Koning J, Ottenkamp J, Schilte PP. (1985). The effect of lowering the hematocrit on polycythemia and thrombocytopenia in congenital cyanotic heart defect. Tijdschr Kindergeneeskd. 53(5): 178-181. PMid:2418530
71. Troost E, Van De Bruaene A, Lampropoulos K et al. (2011). The outcome of Eisenmenger patients with trisomy 21 does not differ from patients without trisomy 21. Acta Cardiol. 66(3): 293-301. https://doi.org/10.1080/AC.66.3.2114128; PMid:21744698
72. Rabin KR, Hitzler J, Rodriguez V et al. (2015). Treatment-related mortality (TRM) in children with Down syndrome (DS) and B-lymphoblastic leukemia (B-ALL): an interim report from the children’s oncology group Trials AALL0932 and AALL1131. Blood. 126: 2502.
73. Ayed W, Gouas L, Penault-Llorca F et al. (2012). Trisomy 21 and cancers. Morphologie. 96(314-315): 57-66. https://doi.org/10.1016/j.morpho.2012.10.001; PMid:23141635
74. Webb D, Roberts I, Vyas P. (2007). Haematology of Down syndrome. Arch Dis Child Fetal Neonatal Ed. 92(6): F503-F507. https://doi.org/10.1136/adc.2006.104638; PMid:17804520 PMCid:PMC2675407
75. Wiedmeier SE, Henry E, Christensen RD. (2008). Hematological abnormalities during the first week of life among neonates with trisomy 18 and trisomy 13: data from a multi-hospital healthcare system. Am J Med Genet A. 146A(3): 312-320. https://doi.org/10.1002/ajmg.a.32107; PMid:18203174
76. Xavier AC, Ge Y, Taub J.W. (2009). Down syndrome and malignancies: a unique clinical relationship. J Mol Diagn. 11(5): 371-380. https://doi.org/10.2353/jmoldx.2009.080132; PMid:19710397 PMCid:PMC2729834
77. Xavier AC, Ge Y, Taub J. (2010). Unique clinical and biological features of leukemia in Down syndrome children. Expert Rev Hematol. 3(2): 175-186. https://doi.org/10.1586/ehm.10.14; PMid:21083461
кафедра психиатрии и наркологии 1СПбГМУ им. И.П. Павлова
Общие сведения
Умственная
отсталость – врожденное или приобретенное в раннем детстве (до 3 лет) состояние
общего недоразвития психики с выраженной недостаточностью интеллектуальных
способностей.
Умственная
отсталость может быть обусловлена различными этиологическими и
патогенетическими факторами, действующими во время внутриутробного развития,
родов или в первые годы жизни. В большинстве случаев умственная отсталость
является не болезненным процессом, а патологическим состоянием, результатом
когда-то подействовавшей вредности, и не имеет тенденции к прогредиентности (прогрессированию).
Долгое время
общепринятым термином для обозначения состояний умственной отсталости был
термин «олигофрения» (греч.: oligos – малый, phren – разум, т.е.
малоумие), который предложил Э. Крепелин (1915) для разграничения обозначаемого
им врожденного слабоумия от слабоумия приобретенного (деменции).
Распространенность умственной отсталости
По различным
оценкам распространенность умственной отсталости колеблется от 0,5% до 3%
населения, при этом легкие формы интеллектуальной недостаточности встречаются
чаще тяжелых. Мужчины страдают умственной отсталостью чаще женщин.
Классификация умственной отсталости
Существуют
разные подходы к классификации состояний умственной отсталости. Наиболее
распространенной клинической классификацией является разделение умственной
отсталости по степени интеллектуального дефекта. Традиционно выделялось три
степени умственной отсталости: дебильность, имбецильность и идиотия. В
Международной Классификации Болезней 10-го пересмотра (МКБ-10) умственная
отсталость представлена в отдельной рубрике (F7) и
подразделяется по тяжести на четыре степени: легкую (F70), умеренную
(F71), тяжелую (F72) и глубокую
(F73). Другая клиническая классификация
предполагает подразделение состояний умственной отсталости (независимо от
глубины психического недоразвития) на стеническую, дисфорическую, астеническую
и атоническую формы (Д.Н. Исаев). Кроме того, выделяют «ядерные» формы
умственной отсталости (Н.И. Озерецкий), для которых свойственна тотальность
психического недоразвития, затрагивающая всю психику в целом, и атипичные
формы, для которых свойственна неравномерная структура психического дефекта с признаками
парциального психического недоразвития.
По
этиопатогенезу состояния умственной отсталости разделяют на три основные группы
(Г.Е. Сухарева):
1.
состояния, обусловленные наследственными
(генными и хромосомными) заболеваниями. К этой группе
относят: синдромы Дауна, Клайнфелтера, Тернера, Мартина-Белл, истинную
микроцефалию, энзимопатические формы, связанные с наследственными обменными
нарушениями (фенилкетонурия, галактозурия и пр.), наследственные неврологические
и нервно-мышечные заболевания с умственной отсталостью.
2.
состояния, вызванные действием различных
вредностей во время внутриутробного развития (эмбриопатии и фетопатии). Сюда относят
состояния, вызванные внутриутробными инфекциями (вирусы краснухи, гриппа,
паратита, цитомегаловирус, возбудители сифилиса, токсоплазмоза и пр.),
интоксикациями (например, алкогольной), гемолитической болезнью плода и пр.
3.
состояния, вызванные действием различных
вредностей во время родов или в первые месяцы и годы жизни. Выделяют
умственную отсталость, связанную с родовой травмой и асфиксией в родах, с
черепно-мозговыми травмами и нейроинфекциями, перенесенными в раннем детстве.
Во многих
случаях достоверно выявить этиологические причины умственной отсталости не представляется
возможным, поэтому такие состояния обозначают как недифференцированные
формы. В свою очередь дифференцированные формы умственной отсталости представляют
собой нозологически самостоятельные заболевания с установленной этиопатогенезом
и характерной клиникой. Часто при дифференцированных формах умственная
отсталость является лишь одним из симптомов в ряду других тяжелых проявлений
этих заболеваний.
Клинические проявления и динамика умственной
отсталости
Психические
расстройства при умственной отсталости, как уже отмечалось выше, полиморфны по
характеру и степени выраженности.
Выраженность интеллектуального
дефекта.
По степени выраженности недостаточности интеллектуальных
способностей в МКБ-10 выделяется:
1.
Глубокая
умственная отсталость (идиотия).
При идиотии значительно ограничены
познавательные способности: больные практически не способны понимать обращенную
к ним речь, не узнают людей, ухаживающих за ними (например, мать), не отличают
съедобного от несъедобного (могут поедать несъедобные предметы), не имеют
представлений о пространственных отношениях (например, о высоте: могут падать с
большой высоты), редко формируют представления о горячем, остром и пр. (могут
получать повреждения, ожоги). Большинство больных не в состоянии освоить даже
простейшие навыки самообслуживания (одеться, умыться, пользоваться столовыми
приборами и пр.). Речь или совсем не сформирована (такие больные издают лишь нечленораздельные
звуки) или состоит из нескольких простейших слов. Значительно недоразвиты моторные
функции больных, в связи с чем многие из них не могут самостоятельно стоять и
ходить, передвигаются ползком. Поведение в одних случаях отличается вялостью,
малоподвижностью, в других – склонностью к однообразному двигательному
возбуждению со стереотипными движениями (раскачивание туловищем, взмахи руками,
хлопанье в ладоши), а у некоторых больных с периодическими проявлениями
агрессии и аутоагрессии (могут внезапно ударить, укусить окружающих, царапать
себя, наносят себе удары и т.п.). В большинстве случаев имеют место грубые
неврологические нарушения и тяжелые соматические аномалии. Жизнь таких больных,
нуждающихся в постоянном уходе и надзоре окружающих, определяется
удовлетворением простейших жизненных потребностей. Коэффициента умственного
развития (стандартизированная методика Д. Векслера для измерения интеллекта) у
лиц с глубокой умственной отсталостью ниже 20.
2. Тяжелая
умственная отсталость (тяжелые варианты имбецильности)
Познавательная
деятельность ограничена возможностью формировать только простейшие
представления, абстрактное мышление, обобщения больным недоступны. Больные
овладевают лишь элементарными навыками самообслуживания, их обучение
невозможно. Словарный запас ограничен одним-двумя десятками слов, достаточных
для сообщения о своих основных потребностях, выражены дефекты артикуляции. Часто
присутствуют неврологические расстройства, нарушения походки. Больные нуждаются
в постоянном контроле и обслуживании. Коэффициент умственного развития этих
пациентов находится в пределах 20-34.
3. Умеренная умственная
отсталость (варианты имбецильности легкой и средней степени)
Эти больные
способны образовывать большее число и более сложные представления, чем больные
тяжелой умственной отсталостью. Больные овладевают навыками самообслуживания,
могут быть приучены к простейшему труду путем тренировки подражательных
действий. Их словарный запас богаче, они в состоянии изъясняться простыми
фразами, поддерживать простую беседу. Относительная адаптация больных с
умеренной умственной отсталостью возможна лишь в хорошо знакомых им условиях,
любое изменение ситуации может поставить их в затруднительное положение из-за
невозможности перехода от конкретных, полученных при непосредственном опыте,
представлений к обобщениям, позволяющим переносить имеющийся опыт в новые
ситуации. Больные не могут жить самостоятельно, нуждаются в постоянном
руководстве и контроле. Некоторые из них могут выполнять простейшую работу в
специально созданных условиях (например, в лечебно-трудовых мастерских).
Коэффициент умственного развития этих пациентов находится в пределах 35-49.
4. Легкая
степень умственной отсталости (дебильность).
Познавательные
расстройства у этих больных заключаются в затруднении формирования сложных
понятий и обобщений, невозможности или затруднении абстрактного мышления.
Мышление у них преимущественно конкретно-описательное, достаточно развита
обиходная речь. Больные легкой степенью умственной отсталости способны к
усвоению специальных программ, основанных на конкретно-наглядном обучении,
которое проводится в более медленном темпе, а также способностью к овладению
несложными трудовыми и профессиональными навыками. Благодаря относительно более
высокому, чем при других степенях умственной отсталости, темпу психического
развития у больных с дебильностью во многих случаях возможна удовлетворительная
адаптация к обычным условиям жизни. Часто эти больные обнаруживают хорошую
практическую осведомленность («их умения больше их знания» — Э. Крепелин). Многие
больные с легкой умственной отсталостью заканчивают специализированные школы и
профессиональные училища, продуктивно работают, заводят семьи, самостоятельно
ведут хозяйство. По сравнению с другими степенями олигофрении черты личности и
характера больных отличаются большей дифференцированностью и индивидуальностью.
Однако эти больные с трудом формируют собственные суждения, но легко перенимают
чужие взгляды, иногда попадая под нездоровое влияние окружающих (например,
могут вовлекаться в бредовые переживания психически больных с формированием
индуцированного бреда, или становиться орудием в руках злоумышленников,
манипулирующих ими для получения собственной выгоды). Коэффициент умственного
развития этих пациентов находится в пределах 50-69.
Эмоционально-волевые
нарушения
Интеллектуальная несостоятельность – самое яркое проявление
умственной отсталости, но она является лишь частью общего психического
недоразвития личности. При олигофрениях значительно страдают эмоциональные и волевые
процессы. Д.Н. Исаев, по особенностям нарушений эмоционально-волевой сферы, вне
зависимости от степени выраженности психического недоразвития, выделяет
следующие формы умственной отсталости:
1.
Стеническая. Волевые процессы у этих больных обладают достаточной
силой и устойчивостью. Больные работоспособны, деятельны. При легких степенях
интеллектуальной недостаточности они имеют хорошую способность к адаптации,
способны в полной мере использовать усвоенные навыки и знания. У некоторых
больных отмечается аффективная неустойчивость, поэтому выделяют два варианта
стенической формы: уравновешенный и неуравновешенный.
2.
Дисфорическая. Характеризуется постоянным злобно-тоскливым аффектом,
склонностью к дисфориям, импульсивным поступкам, негативизму, конфликтности, расторможенности
влечений. Даже при незначительной интеллектуальной недостаточности такие больные
неспособны к обучению и труду. Во время дисфорий они часто проявляют агрессию
(обычно по отношению к близким, осуществляющим за ними уход, при этом агрессия
может быть очень жестокой и изощренной) и аутоагрессию (наносят себе глубокие
порезы, прижигают кожу сигаретой, вырывают волосы и пр.).
3.
Астеническая. Характеризуется нестойкостью волевых процессов, быстрой
истощаемостью, утомляемостью, медлительностью, нарушениями внимания,
затруднениями в усвоении и использовании практических навыков.
4. Атоническая.
Характеризуется практически полным отсутствием способности к психическому
напряжению и целенаправленной деятельности. Больные или полностью бездеятельны
или находятся в состоянии хаотической двигательной расторможенности.
Динамика
умственной отсталости
В большинстве случаев состояния умственной
отсталости относительно стабильны («непрогредиентны»). Однако, иногда, под
влиянием внутренних и внешних факторов, отмечается их положительная или отрицательная
динамика. При своевременном и активном проведении лечебно-коррекционных и
воспитательных мероприятий большинство больных, страдающих легкой и умеренной
умственной недостаточностью, оказываются способны к труду. В процессе
возрастной эволюции и под влиянием лечебных мероприятий отмечается редукция двигательной
расторможенности, импульсивности, негативизма, астенических состояний и пр.
Отрицательная динамика умственной отсталости возможна при присоединении
дополнительных патогенетических механизмов поражения головного мозга (например,
отложение амилоида при болезни Дауна), при действии дополнительных внешних
вредностей (черепно-мозговые травмы, алкоголизм и пр.), психогениях,
неблагоприятном социальном окружении, в периоды возрастных кризов и пр. Декомпенсации
при умственной отсталости могут проявляться цереброастеническими и
психопатоподобными расстройствами, психозами с помрачением сознания, галлюцинаторно-бредовыми,
аффективными психозами и пр.
Дифференцированные формы умственной
отсталости
Состояния,
обусловленные наследственными (генными и хромосомными) заболеваниями
Синдром Дауна
Обусловлен
трисомией по 21 хромосоме. Впервые был описан английским врачом Дж. Дауном в
1866, но связь между нарушением числа хромосом и клиническими проявлениями
болезни была установлена лишь в 1959 году (Ж. Лежен). Частота рождения детей с
синдром Дауна приблизительно 1:700, однако в настоящее время, в связи с
возможностями пренатальной диагностики, есть тенденция к ее снижению. Наиболее
важным фактором риска данной хромосомной аберрации признается возраст матери
(больше 35 лет).
Клинические
проявления: Умственная отсталость при синдроме Дауна может быть
выражена по-разному, чаще это умеренная и тяжелые степени, реже легкая. У
больных отмечается позднее появление и выраженное недоразвитие речи (недостаточное
понимание речи, бедный словарный запас, дизартрия). Дети с болезнью Дауна
обычно не способны к обучению даже по программе вспомогательной школы и
нуждаются в индивидуальном обучении. Эмоциональная
сфера остается достаточно сохранной: большей частью больные ласковы, послушны,
привязаны к родителям, приветливы, добродушны («солнечные дети»), хотя бывают
упрямы. Многие из них любопытны и обладают хорошей подражательной способностью,
что способствует развитию навыков самообслуживания и несложных трудовых процессов.
Редко встречаются безразличные и злобные больные. Как правило, дети с болезнью
Дауна лучше развиваются в родительских семьях, чувствуя заботу близких, но, даже
в этом случае, они не достигают удовлетворительного уровня социальной адаптации
и нуждаются в постоянной опеке.
Особенностью
возрастной динамики болезни Дауна является позднее половое созревание и раннее
появление признаков инволюции (в 30-40 лет). При синдроме Дауна отмечается высокая
частота раннего развития (после 35 лет) атрофических изменений коры головного
мозга и накопления амилоида в виде сенильных бляшек, т.е. морфологические
изменения сходные с болезнью Альцгеймера. При этом больные быстро утрачивают обыденные
навыки, словарный запас, становятся бездеятельными, безразличными, появляются
неврологические расстройства.
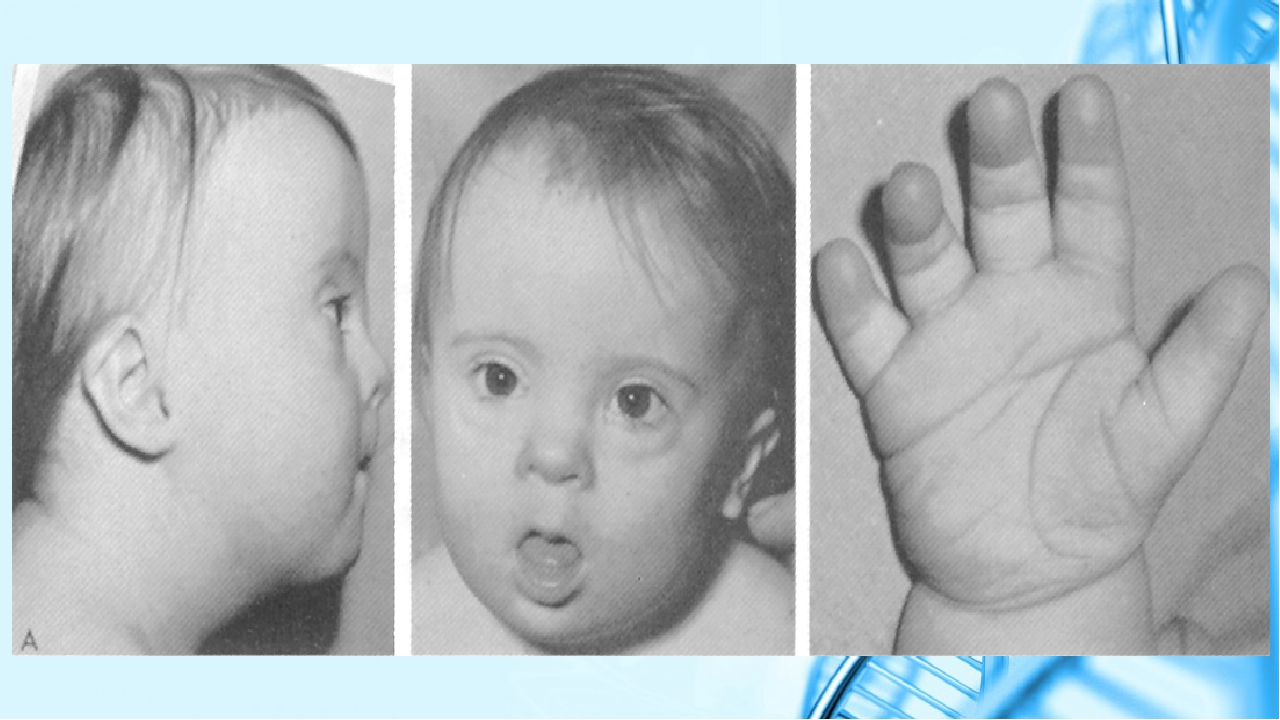
Больным с
синдромом Дауна свойственен специфический физический фенотип, определяемый
множеством стигм дизонтогенеза, что делает возможным диагностику этого
состояния уже при рождении. Дети обычно небольшого роста; при рождении часто
имеют низкую массу тела (до 2500
г). Окружность головы уменьшена. Лицо плоское,
монголоидный разрез глаз (первое название синдрома «монголизм»), широкая
переносица, нос короткий, изредка наблюдается пятнистая окраска центральной
части радужки; нередки катаракты. Язык большой, исчерченный, рот полуоткрыт.
Уши небольшие, прилегающие. Кисти широкие, пальцы короткие, на ладонной
поверхности кисти поперечная борозда. Часто встречаются патология строения
сердечно-сосудистой системы, эндокринные нарушения (гипофункция щитовидной
железы, гипофиза, надпочечников, половых желез), мышечная гипотония. Характерна
повышенная восприимчивость к инфекциям. Продолжительность жизни больных
значительно снижена, не более 10% живут более 40 лет.
Синдром
Тернера (Шерешевского-Тернера)
Моносомия Х-хромосомы (45, Х0). Распространенность
1:3300 новорожденных.
Клинические
проявления: Умственное недоразвитие обнаруживается только у части
больных; обычно легкой степени. Больные трудолюбивы и благодушны. У многих
больных есть критика к своему состоянию и переживание дефекта, отмечается
склонность к невротическим реакциям
Врожденные
аномалии строения придают больным своеобразный вид: низкий рост (как правило,
не превышает 150 см),
диспропорциональное телосложение (преобладание верхней части туловища, широкие
плечи, узкий таз, укорочение нижних конечностей, конституция приближается к
мужской). Шея короткая с избыточной кожей на заднебоковой поверхности, которая
у многих больных выступает в виде шейной складки. Выявляются признаки полового
инфантилизма (наружные половые органы недоразвиты, молочные железы не развиты,
соски втянуты, оволосение лобка и подмышечных впадин отсутствует или скудное). Характерными
признаками являются первичная аменорея, аномалии строения внутренних половых
органов.
Синдром
Клайнфелтера
Дисомия по Х хромосоме у мужчин (47, ХХY). Распространенность
1:1400 новорожденных (мальчики).
Клинические
проявления: Умственная отсталость встречается примерно у четверти
больных, преимущественно в легкой степени. Отмечается выраженная незрелостью
эмоционально-волевой сферы. У многих больных часто присутствует сознание своей
неполноценности, которое становится источником внутреннего конфликта, характерны
невротические и патохарактерологические реакции. Описаны случаи с депрессивными,
ипохондрическими, навязчивыми, шизофреноподобными расстройствами
Внешние вид
больных: характерен высокий рост, астеническое сложение, узкие плечи,
удлиненные конечности, слабо развитая мускулатура. Постоянными признаками
синдрома Клайнфелтера являются недоразвитие половых органов и бесплодие. Примерно
у половины больных отмечается гинекомастия и евнухоидные признаки. В
неврологическом статусе в ряде случаев имеются мышечная гипотония и
диэнцефально-вегетативные расстройства по типу панических атак.
Умственная
отсталость, вызванная наследственными дефектами метаболизма
Фенилпировиноградная
умственная отсталость (фенилкетонурия, ФКУ, болезнь Феллинга)
— наследственное нарушение обмена веществ (характеризуется
аутосомно-рецессивным типом наследования), обусловленное дефицитом одного из
ферментов обмена аминокислоты фенилаланина, что приводит к нарушениям окисления
фенилаланина в тирозин, недостаточным синтезом катехоламинов (адреналина и
норадреналина), гормонов щитовидной железы, меланина, серотонина. В результате
в организме происходит постепенное накопление фенилаланина и его метаболитов,
оказывающих токсическое действие на ЦНС, формируется дефицит гормонов и
медиаторов нервной системы с дальнейшей задержкой психического развития.
Фенилаланин и его метаболиты (фенилкетоновые вещества) выделяются с мочой. Выявляются
значительные этнические различия в распространенности фенилкетонурии. В России
частота среди новорожденных составляет 1:6-10 тыс.
Клиническая
картина: Дети, больные фенилкетонурией, рождаются с нормально
сформированным и функционально полноценным головным мозгом (так как
биохимические процессы плода обеспечиваются обменом веществ матери).
Биохимические нарушения начинают развиваться сразу после рождения. Уже в 4-6 мес.
выявляется отставание в психомоторном развитии, которое заметно прогрессирует. Развернутая
клиническая картина заболевания включает умственную отсталость тяжелой или
глубокой степени, нарушения поведения и кататонические расстройства (состояния
психомоторного возбуждения, импульсивные действия, стереотипные движения,
эхопраксия, эхолалия, субступорозные состояния), астенические состояния. Часто
обнаруживается повышение мышечного тонуса, судороги (у 30% больных), гиперкинезы,
тремор пальцев рук, атаксия, нарушения координации, энурез. Характерны дефекты
пигментации (большинство больных — блондины, со светлой, лишенной пигмента
кожей и голубыми глазами). Моча имеет своеобразный запах («запах волка»,
«мышиный», «затхлый»). Биохимическая диагностика фенилкетонурии основывается на
положительной реакции мочи с FeCl3 на фенилпировиноградную кислоту (проба Феллинга)
и обнаружении повышенной концентрации фенилаланина в плазме крови.
Фенилкетонурия
пример наследственного заболевания с возможностью хорошего эффекта при
своевременной профилактической терапии: для предотвращения развития психических
и неврологических расстройств с первых месяцев жизни и до 10-12 летнего
возраста используются диеты с резким ограничением фенилаланина (полностью
исключают животный белок, значительно растительный, дефицит белков компенсируют
специальными смесями аминокислот без фенилаланина). Чувствительность нервной
ткани к токсическому влиянию продуктов обмена фенилаланина, а также к другим
нарушениям обмена наиболее высока в раннем возрасте (в период созревания мозга).
После окончания процесса миелинизации повышение фенилаланина в крови уже не
оказывает патогенного воздействия на мозг.
Умственная
отсталость, вызванная действием вредностей во время внутриутробного развития
(эмбриопатии и фетопатии)
Умственная отсталость,
вызванная вирусом краснухи (рубеолярная эмбриопатия). При
заболевании беременной краснухой в первом триместре беременности формируется
эмбриопатия с грубыми нарушениями строения нервной системы (микроцефелия,
порэнцефалия), органов слуха и зрения, врожденными пороками внутренних органов.
Умственная отсталость у таких больных обычно глубокой степени, часты судорожные
припадки.
Умственная
отсталость, обусловленная гемолитической болезнью плода и новорожденного. Гемолитическая
болезнь плода (эритробластоз плода) обусловлена резус-конфликтом между матерью
и ребенком, который приводит к гемолизу эритроцитов плода, анемии, высокому
уровню билирубина, нарушениям кровообращения, отекам, повышению внутричерепного
давления. Одним из последствий этого состояния может быть формирование
умственной отсталости, выраженность которой бывает различной.
Умственная
отсталость, обусловленная алкоголизмом матери (алкогольная фетопатия). Развивается
при употреблении алкоголя матерями во время беременности. По данным ряда
авторов занимает первое место среди причин легкой умственной отсталости. Клиническая
картина складывается из умственной отсталости (преимущественно легкой степени),
задержки физического развития (особенно выраженная при рождении и в первые годы
жизни), нарушения строения черепа (микроцефалия, укорочение глазных щелей,
недоразвитие костей срединной части — выпуклый лоб, короткий нос с широким и
плоским переносьем, гипоплазия верхней челюсти).
Состояния,
вызванные действием вредностей во время родов или в первые месяцы и годы жизни
Умственная
отсталость, обусловленная родовой травмой или асфиксией в родах. Механическое
повреждение черепа ребенка во время родов может приводить к внутричерепным
кровоизлияниям или даже к непосредственным повреждениям мозга и его оболочек.
Кислородное голодание вызывает нарушения метаболизма в нервной ткани. Эти факторы
могут приводить к формированию органической патологии головного мозга и, в
последующем, умственной отсталости (выраженность которой может быть различна).
Для родовых травм характерны очаговые неврологические расстройства, судорожные
припадки, иногда – гидроцефалия.
Умственная
отсталость, обусловленная нейроинфекциями, перенесенными в раннем детстве. После
перенесенных менингитов и менингоэнцефалитов может формироваться умственная
отсталость различной степени выраженности, очаговые неврологические
расстройства, судорожные припадки, гидроцефалия.
Дифференциальный диагноз
Диагноз
умственной отсталости основывается на установлении психического дефекта, центральное
место в котором занимает недоразвитие интеллектуальных способностей, на
обнаружении признаков отставания в психическом развитии в детском и
подростковом возрасте, а также отсутствии прогредиентности, т.е. признаков
углубления психического дефекта. С целью определения степени интеллектуального
дефекта используют специальные психологические методы оценки интеллекта (см.
главу 7). Для уточнения этиологии некоторых форм умственной отсталости
требуются дополнительные лабораторные и инструментальные исследования.
Дифференцировать умственную отсталость необходимо с состояниями, обусловленными
педагогической запущенностью (случаи, когда здоровый ребенок лишен условий для
правильного умственного развития) и прогредиентными психическими заболеваниями
(в первую очередь с шизофренией и эпилепсией, манифестирующими в раннем детском
возрасте).
Прогноз
Прогноз умственной
отсталости зависит от степени недоразвития психики, выраженности
интеллектуального дефекта, особенностей эмоционально-волевой сферы больных,
этиологии. При неосложненной умственной отсталости легкой степени возможна
полная социальная адаптация, которая исключает необходимость в наблюдении
психиатра. В то же время социальный прогноз глубокой и тяжелой степеней умственной
отсталости неблагоприятен.
Биохимия и жизнь
Почти все беременные женщины в Москве проходят биохимический скрининг – тест, который может определить наличие синдрома Дауна у ребенка. Каждая пятая попадает в группу риска.
Корреспондент «Большого города» Светлана Рейтер оказалась одной из них, чуть не сошла с ума и решила выяснить, что это такое и как все это устроено. Мой ребенок может родиться с синдромом Дауна. Так и было написано на бумажке: «Результат скрининга на синдром Дауна — ВЫСОКИЙ РИСК». Этот ВЫСОКИЙ РИСК — именно так, большими буквами отпечатанный — сводил на нет все мое благополучие.
Дружная семья, собака, квартира в центре Москвы, машина, купленная с расчетом на второго ребенка, да и сам непосредственно ребенок — все это разваливалось на глазах, придавленное ВЫСОКИМ РИСКОМ, обнаруженным в результате простейшего анализа — биохимического скрининга по выявлению внутренних пороков развития, который в наше время делает каждая будущая мать. Каждая пятая попадает в группу риска.
Частый случай Биохимический скрининг — единственная неинвазивная (безопасная для жизни ребенка) возможность на ранней стадии заподозрить внутренние пороки развития плода. Такие как дефекты нервной трубки, синдром Эдвардса и самый частый и известный порок — синдром Дауна. При попадании в группу риска у беременной есть три варианта. Делать дальнейшие — инвазивные (вторгающиеся в связку «мать-плод») исследования. Для этого живот прокалывают тонюсенькой иглой и берут частицы пуповинной крови, плаценты и околоплодных вод. Не обращать на результаты теста никакого внимания и рожать, как бы то ни было. Или — сразу идти на аборт.
Инвазивных анализов беременные боятся: риск выкидыша, кровотечений и преждевременных родов при них — 1%. Не каждая согласится. Но мамочки, как их называют в женских консультациях, хотят знать всю правду. Даже самую страшную. Каждый день в лабораторию пренатальной диагностики при Медико-генетическом научном центре приходят женщины, попавшие в группу риска, для дальнейших анализов. Примерно раз в неделю в лаборатории после инвазивных исследований у плода обнаруживают синдром Дауна.
Лабораторией руководит доктор биологических наук, профессор Татьяна Золотухина: «Тридцать лет назад мы делали акцент на исследованиях женщин старшего, опасного репродуктивного возраста. Но, проведя десять лет в таком режиме, мы не получили никакого результата. Детей с синдромом Дауна рождалось столько же. А дело в том, что основную массу детей с синдромом Дауна рожают молодые женщины. Риск рождения ребенка с синдромом Дауна у них ниже, но численно их куда больше. Встал вопрос: «Каким же образом можно заподозрить синдром Дауна у женщин, которые не относятся к группе риска по возрасту?».
В 1984 году британскими учеными Николасом Дж. Уолдом и Говардом Каккелом был предложен искомый неинвазивный, совершенно безопасный биохимический тест по нескольким маркерам крови. У женщин, а также у медиков и генетиков появилась возможность «цеплять» синдром Дауна на той ранней стадии, когда еще можно принять решение — оставлять плод или избавляться от него без явного ущерба для здоровья беременной (согласно нашему законодательству, женщина может прервать беременность по собственному желанию только до двенадцати недель. Прерывание же на поздних сроках— до 22-й недели — возможно только по медицинским показаниям, угрожающим жизни матери или плода; диагностированный синдром Дауна у плода — одно из них).
Единственное но: стандартная беременная не знает, что биохимический скрининг на выявление пороков развития — метод вероятностный. Это ни в коем случае не диагноз, скрининг лишь помогает составить группу риска. Точность его — 60%.
Мировой опыт. В США скрининг добровольный, рекомендован женщинам старшего репродуктивного возраста или с патологиями в семейном анамнезе. И любой врач-гинеколог объясняет беременной, что скрининг — метод вероятностный и точных гарантий не дает. Делается это для того, чтобы как-то минимизировать стресс, которому так или иначе подвергается любая беременная, попавшая в группу риска.
Чтобы как-то увеличить точность, в лаборатории Золотухиной впервые в мире предложили одновременно с биохимическим скринингом на тех же сроках проводить УЗИ: известно, например, что у детей с синдромом Дауна часто увеличена шейная складка и уменьшена носовая кость. Точность теста увеличивается до 85%. В 15% попадают те здоровые дети, у которых увеличена шейная складка и уменьшена носовая кость. И наоборот.
Результат теста. Несложную процедуру скрининга я, как положено, проходила на одиннадцатой неделе; врач женской консультации № 6 на Маросейке Светлана Шамаева выдала мне направление, а потом позвонила и сказала: «Все нормально». И я успокоилась. На следующий день мне позвонила мама и сказала, что результаты от нормы далеки. И врач не хотела говорить мне об этом прямо, чтобы не расстраивать. И надо срочно идти в женскую консультацию, чтобы получить направление к генетику. А я еще помню, что раньше врачи так себя вели только в самых ужасных случаях: пациенту говорили, что он здоров, а родственникам — что у него рак в последней стадии.
Врачебная ошибка. Коридор возле кабинета генетика в Центре планирования семьи и репродукции ежедневно с утра до вечера заполнен женщинами, нервно сжимающими в руках клочок бумаги. Такой же, как у меня. С таким же ВЫСОКИМ РИСКОМ. На точность анализа влияют гинекологические проблемы, диабет, проблемы печени, неправильно рассчитанный срок беременности, вес и возраст беременной. «Послушай, — сказал мне муж, — ты посмотри, сколько здесь сидят женщин. Все в группе риска. Ждут генетика. Неужели ты думаешь, что у всех у них родятся дауны?». Мой муж — голландец. Они умеют хранить выдержку в любых, пусть и самых патовых ситуациях. На шестнадцатой неделе мне сделали один из инвазивных тестов — амниоцентез. Велели ждать ответа. Ну а потом старшая дочь бегала по квартире и кричала: «У мамы ребенок нормальный!». Так оно и получилось. Так часто получается: всего семь женщин из ста, попавших в группу риска, носят плод с пороками развития. Девяносто три волнуются зря. Но очень сильно. Будущее Золотухина честно говорит, что синдром Дауна настолько коварен, что мы не можем с уверенностью дать какие-то гарантии. Результаты биохимического скрининга в комбинации с возрастом женщины и результатами УЗИ вносятся в программу «Прогноз», разработанную в лаборатории Золотухиной, и на выходе женщина имеет цифры. На точность анализа влияют проблемы по гинекологической части или и вовсе диабет и нарушения работы печени. Неправильно рассчитанный срок беременности, неточно указанный вес беременной, ее возраст — тоже могут привести к ложноположительным и ложноотрицательным результатам.
Более четкой альтернативы этому методу нет — правда, в государстве Израиль система дородовой диагностики строится по несколько отличному принципу: здесь почти все женщины старшего репродуктивного возраста делают инвазивные исследования, а именно забор околоплодных вод — амниоцентез. Как и биохимический тест в Москве, амниоцентез в Израиле — бесплатная процедура. Поскольку амниоцентез проводится в большом количестве израильских клиник и методика его отработана, риск самопроизвольного выкидыша снизился с одного процента до 0,5%. Вероятно, когда-нибудь все будет гораздо проще. Генетиками, по словам Руфи Шомрат, профессора института генетики при медицинском центре «Ихилов» в Тель-Авиве было совершено открытие: оказывается, во время беременности в периферическую кровь матери попадают клетки плода. Значит, в крови матери можно обнаружить его, плода, ДНК и РНК. Если найти способ извлекать эти клетки из крови матери, появится возможность прийти к новому диагностическому подходу. И это уже будет стопроцентный диагноз.
Путаница. Кровь на биохимический скрининг по выявлению у плода внутренних пороков развития в последние три года берут в любой женской консультации; действуют согласно приказу департамента здравоохранения номер 144: «О совершенствовании организации пренатальной диагностики врожденной и наследственной патологии», подписанному руководителем департамента Андреем Сельцовским 4 апреля 2005 года. С этого момента скрининг в комбинации с УЗИ стал обязателен, насколько вообще обязательны походы в женскую консультацию. (Можно, безусловно, ими пренебречь; но вряд ли Вы сможете без постановки на учет в консультацию попасть в желанный роддом. Хотя формально Вам не могут отказать в родовспоможении, даже если в обменной карте вместо результатов скрининга у вас будет записан отказ). Ходишь в консультацию — значит, сдавай кровь на биохимический скрининг. Звоню в 15-ю женскую консультацию: «Биохимический тест на пороки развития беременным у вас обязательный?». «Ну конечно!». То же самое говорят в консультациях № 12 и № 7. Заведующая женской консультацией при 20-й поликлинике Людмила Конышкина объясняет: «Беременные у нас послушные. Им сказали делать — они и делают. Мы в консультации сами берем кровь, сами отвозим в лабораторию. На все берем — на сифилис, СПИД и заодно на скрининг. У всех без исключения, независимо от возраста, согласно приказу номер 144. Если высокий риск — направляем на консультацию к генетику».
Многочисленные противники биохимического скрининга утверждают, что подобные анализы толкают женщин к так называемым, евгеническим абортам. Статистически такие аборты нигде не фиксируются — просто, потому что прерывая беременность на ранней стадии, женщина имеет право не указывать причину. Результаты же биохимического скрининга показанием к аборту не являются. Но вот письмо некоего Руслана, опубликованное на сайте швейцарской клиники Swissmed: «Моя жена сделала скрининг-тест на какую-то патологию на 12-й неделе беременности. Спустя неделю в 11 утра она получила результаты. Позвонив мне на работу, она поставила меня перед фактом, что ей срочно необходимо сделать чистку и сделала аборт. Правы ли врачи и могу ли я получить результаты теста, могут ли врачи без повторного анализа и в такой краткий срок направить ее на чистку? Как я могу выяснить правду?». И ответ: «Генетический скрининг выявляет риск врожденной патологии плода. Если этот риск высокий, женщина может принять решение о прерывании беременности самостоятельно. Клиники информацию о здоровье пациента не выдают никому, кроме лично пациента». Про «вероятностный метод» ни в ответе на письмо, ни в консультациях перед анализом не объясняют, если специально не спросить. Потому что, как говорит ассистент кафедры акушерства и гинекологии при РАМН имени Сеченова Сергей Тимофеев: «Когда у тебя в очереди сидят человек тридцать, с каждой беременной рассусоливать не будешь».
Пропущенный диагноз. В 2007 году руководитель департамента здравоохранения Андрей Сельцовский официально заявил о сокращении числа новорожденных с синдромом Дауна в Москве: если в 2002 году их родилось 134, то в 2006-м, спустя год после принятия приказа, всего 94. Тогда же было объявлено о сокращении абортов — если в 2000 году их было сделано 57 324, то год спустя — всего 28 502. Анна Шакина родилась с синдромом Дауна в 2006 году. Наверное, статистика учитывает и ее. Сейчас Ане два года. Ее мать, 38-летняя Алла Шакина, педагог-воспитатель детского сада, делала все, как положено. Обследования проходила в Центре акушерства, гинекологии и перинатологии. Все результаты у Аллы были идеальными. «У меня даже не заподозрили ничего: отличные скрининги на двенадцатой неделе, идеальные результаты по УЗИ. Я думала сделать какие-то дополнительные исследования — 38 лет все-таки, опасный возраст, к тому же первый ребенок. Но врачи сказали, что особой нужды нет. Я ведь даже не попала в группу риска». Это непопадание в группу риска Алла воспринимает наиболее тяжело. «Я просто не была хоть как-то готова к тому, что у меня родится ребенок с синдромом Дауна. Если бы я знала заранее, я бы приняла решение, делать аборт или не делать. А тут мне казалось, что это конец. Я не жила с этим. Я после родов погибла сразу». За два дня до родов Алле сказали: «Ну все, мы ждем абсолютно здоровую девочку». Через два дня ей сделали кесарево сечение. Затем она лежала в палате и принимала поздравления. Ей сказали просто: родилась девочка. И все. Когда Алле объявили, что у девочки синдром Дауна, первая мысль ее была: «Немедленно отказаться». Что она и сделала. «Я сказала — отказываюсь. Она мне не нужна. Такая». Потом были беседы с психологом из фонда поддержки детей с синдромом Дауна Downside-Up, уговоры мужа Кирилла, согласного на любого ребенка. Аню забрали домой на девятнадцатый день жизни. «Кто ж виноват, что у меня какие-то клеточки не высеялись? — говорит теперь Алла. — Но если я во второй раз забеременею, то от скрининга наотрез откажусь. Я в него совершенно не верю. Результат не значит ничего. Я сейчас дочь люблю безгранично. Я даже и не знаю, как теперь жить без нее. Я себя гноблю, что позволяла жить без нее девятнадцать дней».
Из четырех женщин, которым по результатам скрининга рекомендовали амниоцентез, через несколько недель в палату пришли только трое.
Ложный диагноз. С ней согласна Наталья Самарина, 39-летняя медсестра: «В 61-й поликлинике мне дали пачку направлений на анализы, сказали: все обязательно сделать. Вот анализ на СПИД/сифилис, вот — общий анализ крови, а вот — биохимический тест, его сейчас все делают. Опять же, обещали, что в случае плохого результата мне обязательно позвонят». Звонка не последовало, и в следующий раз Наталья Самарина пришла в поликлинику уже на довольно солидном сроке, совершенно уверенная в том, что все у нее в полном порядке. «Пожилой врач прямо мне сказала: «Аборт делать не планируете?» А я на сроке 20 недель. Какой аборт? Мне сказали: высокий риск. Я не трепыхнулась. Отказалась от амниоцентеза из-за риска потерять ребенка», — вспоминает Наталья. Наталья Самарина родила девочку Дашу с синдромом Дауна: «Скрининг — бестолковый анализ. Он никому не нужен. Только нервы зря треплет. Он даже вреден, так как толкает к амниоцентезу, а для здоровья это риск гораздо больший, чем риск родить дауненка. Ученые всегда забывают, что рождается все-таки не синдром. Рождается ребенок. Как моя дочка Даша».
От амниоцентеза отказалась в свое время и врач-педиатр «Он-клиник» Марина Терехина, 28-летняя мать четверых детей: «С последним ребенком мне поставили высоченный риск синдрома Дауна. Я пошла на консультацию к генетику в Центр планирования семьи и репродукции, и мне предложили делать амнио-центез. Я сказала: «Слушайте, у меня куча противопоказаний. Давайте бумагу, я напишу отказ». Два года назад у Терехиной родилась здоровая дочь Катя. Мама Нестора Мхеидзе, редактор на телевидении Елизавета Ефимова считает по-другому: «Такой анализ очень нужен. Он дает право самостоятельно принимать решение, что делать дальше. Хуже родить ребенка в неведении, а потом рыдать всю жизнь. На момент беременности мне было 33 года, и я хотела с уверенностью знать, что с ребенком все в порядке. Если бы в итоге у меня обнаружили серьезную внутриутробную патологию, я бы беременность прервала». Рассуждения отечественных адептов биоэтики о том, что биохимические скрининги толкают будущую мать к аборту и что все эти скрининги — от лукавого, то есть от евгеники, сочетавшейся браком с генетическим отбором, в случае с Москвой все-таки неправомерны. Просто потому что многие женщины редко понимают, что они делают, а группа риска — условна. У нас санпросвет заменен интернетом; скрининг стал зловещей городской легендой беременных. В итоге происходит полнейшая чехарда: женщины, попавшие в группу риска и родившие здорового ребенка, утверждают, что все эти анализы — полный бред. Такого же мнения придерживаются те, кто в группу риска не попал, а ребенок родился с патологией. Из четырех женщин, которым одновременно со мной по результатам скрининга рекомендовали амниоцентез, через несколько недель в палату пришли только трое. А четвертая сказала: «Может, сразу сделать аборт?» Главный врач Но есть человек, который знает ответы на все мои вопросы. В его подчинении — все женские консультации города Москвы. Он был одним из тех, кто продвигал введение повсеместного скрининга. Причем, что немаловажно, добивался того, чтобы скрининг был бесплатным для всех беременных. Марк Курцер — это ответ на все молитвы беременной женщины. Главный врач Центра планирования семьи и репродукции и главный акушер-гинеколог города Москвы. Он выгодно отличается от врачей советской складки: молод, хорош собой, разговаривает мало и по делу. Про него рассказывают легенды: безоперационное лечение миомы матки, кесарево сечение в кратчайший срок — он был первым во многих бластях. Он против домашних родов, за внедрение бесплатной программы экстракорпорального оплодотворения и другое. Многие темы волнуют Марка Курцера. Меня же, когда я шла к нему на интервью, интересовала неразбериха со скринингом. Он, судя по всему, один из немногих, кто способен что-то объяснить. В конце концов, он один из тех, на кого возложена ответственность следить за исполнением 144-го приказа (так и написано). Если и есть в этом городе мужчина, который понимает, что испытывает женщина из группы риска, идущая на консультацию к генетику, — это он. В этот день у Марка Курцера прошли две операции. Он был хмур и неприветлив. Потом, правда, с увлечением рассказывал, что любая женщина может пройти бесплатную процедуру ЭКО и что каждая беременная имеет право на родовспоможение в любом понравившемся ей роддоме. Но после вопроса, может ли женщина отказаться от биохимического скрининга, Марк Курцер помрачнел, выдвинул новую версию: «Может. По религиозным соображениям» — и предложил закрыть тему. «Ну а вот как вы посоветуете себя вести женщинам, попавшим в группу риска?» — спросила я. И тут он вспылил — как гений, которому задают неудобные вопросы, подвергая сомнению его гениальную теорию. И я не могу изложить нашу беседу подробным образом. Потому что Марк Курцер вытолкал меня из своего кабинета, вызвав охрану, отобрал диктофон и стер записанное интервью. По дороге домой я думала: как же обидно вышло. Мне хотелось всего лишь понять, кто, когда и как должен объяснять беременным, что результаты биохимического скрининга — не гетто и не приговор. Ведь дело не в том, хочешь ли ты родить ребенка с синдромом Дауна или собираешься делать аборт. Просто, сдавая свою кровь, ты имеешь полное право знать, для чего ты это делаешь. Знания — нет. Права — тоже. А главный гинеколог города Москвы выгнал из своего кабинета женщину, у которой до сих пор среди бумаг валяется листок с цифрами: «Результат скрининга на синдром Дауна — высокий риск». Давно пора его выкинуть. Дело не в том, хочешь ли ты родить ребенка-дауна или сделать аборт. Просто ты имеешь полное право знать, для чего ты это делаешь.
День людей с синдромом Дауна: 5 мифов о «солнечных людях»
Богдан Петришин
21 марта в Украине и мире отмечается Всемирный день людей с синдромом Дауна. Их еще называют «солнечные люди» или «дети солнца» за открытость и искренность.
Впервые этот день был проведен в 2006 году, его инициаторами были Европейская ассоциация Даун-синдром (EDSA) и Всемирная ассоциация Даун-синдром (ІDSA).
В декабре 2011 года Генеральная Ассамблея ООН объявила 21 марта Всемирным днем людей с синдромом Дауна. Март выбран потому, что синдром Дауна представляет собой трисомию (март — третий месяц года) по 21 хромосоме (поэтому и 21 марта).
Синдром Дауна — это генетическая аномалия, которой присуща дополнительная хромосома. Люди с этим синдромом имеют 47 хромосом в кариотипе вместо привычных 46. Синдром известен еще с 1866 года и назван по имени английского врача, который его открыл и описал, но причины возникновения патологии были открыты учеными-генетиками только в ХХ веке.
Синдром Дауна часто приводит к изменениям моторики, физических характеристик и здоровья человека. Жизненно важное значение для развития таких больных имеет доступ к медицинскому обслуживанию, программам раннего вмешательства и инклюзивного образования, а также проведение соответствующих исследований. Несмотря на то, что дети с синдромом Дауна развиваются несколько медленнее, чем обычные, позже начинают ходить и разговаривать — они способны к обучению, многие из них может освоить профессию, достичь оптимального качества жизни и ухаживать за собой без посторонней помощи.
Ежегодно в мире рождается от трех до пяти тысяч детей с этим синдромом, в Украине — более 400. Таких малышей называют еще детьми Солнца, но проблемы, с которыми сталкиваются они и их родители, совсем не солнечные. Если в Англии 80% всех детей с синдромом Дауна учатся вместе с другими школьниками, то в Украине таких наберется чуть процент. Проблемой для этих детей является и посещение детского сада, общественных, культурных учреждений. Обычно украинцы с синдромом Дауна почти обречены на изоляцию и асоциальность. Только в последнее время, благодаря титаническим усилиям родителям детей с синдромом Дауна, которые объединяются для решения общих проблем, и волонтерам ситуация немного сдвинулось с мертвой точки.
5 мифов о людях с синдромом Дауна
1️⃣ Взрослые с синдромом Дауна неработоспособны.
💢 На самом деле: Взрослые с синдромом Дауна во всем мире работают на разных должностях — в банках, корпорациях, гостиницах, больницах, домах престарелых, офисах и ресторанах. Они задействованы в музыкальной и развлекательной индустрии, на официальных должностях, уходе за детьми, спортивном поле и компьютерной индустрии. Как и любой другой, люди с синдромом Дауна хотят устроиться на работу, где их вклад будет цениться.
2️⃣ Люди с синдромом Дауна всегда счастливы.
💢 Насправді: Люди с синдромом Дауна имеют те же чувства, как и любой другой. Они переживают весь спектр эмоций. Неправильное впечатление складывается потому, что люди с синдромом Дауна более открыты и не скрывают своих чувств и реакций. Они так же реагируют на позитив и проявления дружелюбия, но им также больно и грустно от некорректного поведения окружающих.
3️⃣ Ребенок с синдромом Дауна негативно влияет на других детей в коллективе.
💢 На самом деле: Дети с синдромом Дауна прекрасно взаимодействуют с другими детьми в коллективе, они не нуждаются в отдельных заведениях для обучения. Тем более дети с синдромом Дауна не несут никакой угрозы другим детям. Напротив, коллективы, в которых такие дети, более доброжелательные и эмпатические.
В Финляндии, Норвегии, Швеции, Великобритании и других странах дети с синдромом Дауна посещают обычные школы. Пребывание в группе других детей помогает детям с синдромом Дауна чувствовать себя неизолированными. Сегодняшние одноклассники в дальнейшем могут стать коллегами, работодателями. Взаимная социализация полезна для детей, так как им в дальнейшем сосуществовать в обществе.
4️⃣ Дети с синдромом Дауна рождаются только у женщин старше 35 лет.
💢 На самом деле: Вероятность рождения ребенка с синдромом Дауна у женщин после 35 лет выше, но такой ребенок может родиться у женщины любого возраста, даже у 20-летней. Генетический сбой происходит независимо от здоровья, образования, расы, места проживания и социального благосостояния родителей.
5️⃣ Синдром Дауна – это генетическая наследственная болезнь.
💢 На самом деле: Синдром Дауна относится к классу хромосомных аномалий. Поэтому, называть синдром Дауна болезнью — некорректно, правильнее говорить «генетическая хромосомная аномалия». Эта аномалия заключается в том, что в хромосомном наборе человека появляется дополнительная, третья хромосома в 21-й паре, то есть люди с синдромом Дауна имеют 47 хромосом, а не 46, как все остальные. Поскольку синдром Дауна не является болезнью — люди не болеют и не страдают синдромом Дауна. Они просто с ним живут.
Акция «Цветные носки»
Чтобы привлечь внимание к инаковости, однако не неполноценности людей с синдромом Дауна по всему миру в этот день проходит акция «Lots of Socks». Приобщиться к ней просто: надо надеть разноцветные, непарные или просто яркие носки. Таким образом вы продемонстрируете свою солидарность с людьми с нечетным хромосомой, и в то же время обратите на себя внимание прохожих, поймете, как это — быть не таким, как большинство.
Давайте помнить об этих «солнечных людей» и всесторонне поддерживать! Почувствуй себя особенным, помогая особенным! Это еще одно проявление толерантности, который очень необходим нашему обществу.
Болезнь Хантингтона: причины, симптомы, лечение
Что такое болезнь Хантингтона?
Болезнь Хантингтона (БГ) — это наследственное и смертельное заболевание, при котором нервные клетки головного мозга разрушаются . Это приводит к ослаблению физических и умственных способностей, и со временем они ухудшаются. Лекарства нет. Если она начинается в раннем возрасте, это называется ювенильной болезнью Гентингтона.
Диагноз болезни Хантингтона может стать настоящим шоком. Есть много чего принять.
Но обращение к системе поддержки, такой как социальный работник, терапевт или группа поддержки, может сделать путешествие немного менее пугающим. С помощью медицинской бригады люди с болезнью Хантингтона могут жить самостоятельно в течение многих лет.
Симптомы болезни Хантингтона
Хотя симптомы могут впервые проявиться в среднем возрасте, болезнь Хантингтона может поразить любого от детства до пожилого возраста. Симптомы часто впервые появляются в возрасте от 30 до 40 лет. В течение 10–25 лет болезнь постепенно убивает нервные клетки головного мозга.Это влияет на тело, разум и эмоции. Если симптомы проявляются до 20 лет, это называется ювенильной болезнью Хантингтона, и она может ухудшиться быстрее.
Продолжение
Симптомы могут сильно отличаться от человека к человеку. А стресс или волнение могут ухудшить симптомы.
Некоторые симптомы обнаружить легче, чем другие. Ненормальные движения могут быть первым, что вы заметите. Похудание может вызывать беспокойство на всех этапах.
Симптомы болезни Хантингтона имеют тенденцию к развитию поэтапно.
Симптомы на ранней стадии
На ранних стадиях изменения могут быть весьма незначительными, что позволяет продолжать управлять автомобилем и работать. Вам может потребоваться небольшая дополнительная помощь.
Общие ранние симптомы включают:
Проблемы с обучением новому
Проблемы с принятием решений
Провалы памяти
Перепады настроения
Неуклюжесть
Медленные или ненормальные движения глаз
Проблемы с мышцами (дистония)
Проблемы со сном (бессонница)
Потеря энергии и усталость
Симптомы средней стадии
Со временем симптомы начинают больше мешать вашему дню- сегодняшняя жизнь.Например, вы можете начать ронять предметы или падать. Или у вас могут быть проблемы с речью или глотанием.
Сохранять организованность может быть сложно. А эмоциональные изменения могут оказать давление на отношения.
Общие симптомы средней стадии включают:
Неконтролируемые подергивания (хорея)
Проблемы при ходьбе
Путаница
Потеря памяти
Изменения личности
Изменения речи
Проблемы с глотанием
Проблемы с дыханием
Мысли о смерти, смерти или самоубийстве
Потеря веса
Развитие обсессивно-компульсивного расстройства (ОКР), биполярное расстройство расстройство, или мания
Снижение мыслительных способностей
Продолжение
Симптомы поздней стадии
На этой стадии люди с болезнью Хантингтона должны полагаться на заботу других.Невозможно ходить и говорить. Скорее всего, вы все еще будете знать о близких, которые вас окружают. Суетливые движения могут стать серьезными или исчезнуть.
Симптомы ювенильной болезни Гентингтона
У детей и подростков болезнь Хантингтона может прогрессировать быстрее и вызывать такие симптомы, как:
Диагностика болезни Гентингтона
Семейный анамнез играет важную роль в постановке диагноза. Ваш врач задаст вам вопросы о вашем медицинском опыте и проведет медицинский осмотр.Если вы и ваш врач подозреваете болезнь Хантингтона, невролог проведет дополнительные анализы.
Невролог может проверить:
Рефлексы
Мышечная сила
Баланс
Чувство осязания
Зрение
Слух
6 Настроение и психическое состояние
Память
Рассуждение
Навыки мышления
Речь
Лечение и домашние средства от болезни Хантингтона
На данный момент лечение Хантингтона включает в себя управление симптомами:
Лекарства могут помочь контролировать движения.Ваш врач может тесно сотрудничать с вами, чтобы контролировать любые побочные эффекты и при необходимости менять лекарства.
Некоторые антипсихотические препараты обладают побочным эффектом, контролирующим движение, и помогают некоторым людям.
Антидепрессанты также могут помочь при обсессивно-компульсивном расстройстве.
Лекарства, стабилизирующие настроение, могут облегчить симптомы расстройств настроения, но могут вызывать другие побочные эффекты.
Речевая или языковая терапия может быть полезна при любых проблемах с речью или глотанием.
Трудотерапия или физиотерапия могут помочь вам научиться лучше контролировать движения. А вспомогательные устройства, такие как поручни, могут помочь вам управлять изменяющимися физическими способностями.
Нутриционная поддержка варьируется от использования специальной посуды и сосредоточения внимания на продуктах с высоким содержанием питательных веществ до дополнения кормлением через зонд на более поздних этапах.
Упражнение может быть очень полезным. Люди с болезнью Хантингтона, которые остаются в хорошей форме и активны, как могут казаться, добиваются большего успеха, чем те, кто этого не делает.
Психотерапия научит вас управлять изменениями в своих эмоциях и мышлении. Такие стратегии, как разбиение задач на более простые шаги, могут значительно облегчить эти изменения для вас и вашей семьи.
Члены семьи могут помочь, внося некоторые изменения в дом:
Подавайте дополнительное питание и добавляйте высококалорийные добавки, чтобы поддерживать здоровый вес.
Не отвлекайтесь во время еды.
Выбирайте продукты, которые легче пережевывать и глотать.
Используйте вилки и другие принадлежности, предназначенные для людей с ограниченной моторикой.
Используйте закрытые чашки с трубочкой или носиком для питья.
Соблюдайте обычный распорядок дня.
Используйте телефонные или компьютерные напоминания для задач.
Сохраняйте жизнь как можно более спокойной, простой и спокойной.
Для детей, вместе со школьным консультантом составьте план обучения.
Общайтесь с друзьями и поддерживайте социальное взаимодействие как можно чаще.
По возможности добавьте в дом пандусы и лифты для инвалидных колясок.
Добавьте защитные дуги в ванных комнатах, рядом с кроватью и на лестницах.
Используйте голосовое управление освещением и другие функции «умного» дома.
Используйте электронные речевые программы или графические таблицы для облегчения общения.
Причины болезни Хантингтона
В 1993 году исследователи обнаружили ген, вызывающий болезнь Хантингтона.У всех есть ген HD, но в некоторых семьях аномальная копия гена передается от родителя к ребенку. Если у вас есть родитель с болезнью Гентингтона, у вас есть 50% шанс получить этот ген и заболеть.
Также:
Мужчины и женщины с одинаковой вероятностью унаследуют аномальный ген.
Если у вас нет аномального гена, вы не сможете заразиться геном Хантингтона или передать его своим детям.
Болезнь не пропускает поколения.
Если вы или члены вашей семьи планируете пройти тестирование на болезнь Хантингтона, рекомендуется сначала получить профессиональную генетическую консультацию. Консультанты могут помочь объяснить, чего ожидать от результатов теста.
Зная о гене HD, ученые смогли многое узнать о том, как болезнь влияет на мозг. Что еще более важно, это открытие может помочь подготовить почву для будущего лечения.
Генетическая связь с болезнью Паркинсона
Если у вас есть члены семьи, болеющие болезнью Паркинсона, или если вы сами страдаете этой болезнью и беспокоитесь о шансах ее развития у ваших детей, вы, вероятно, уже задавались вопросом: существует ли ген, вызывающий болезнь Паркинсона? Насколько прямая ссылка?
Около 15 процентов людей с болезнью Паркинсона имеют семейный анамнез этого заболевания, и связанные с семьей случаи могут быть результатом генетических мутаций в группе генов — LRRK2, PARK2, PARK7, PINK1 или гене SNCA (см. Ниже).Однако взаимодействие между генетическими изменениями или мутациями и индивидуальным риском развития болезни до конца не изучено, — говорит Тед Доусон, доктор медицинских наук, директор Института клеточной инженерии при Джонсе Хопкинсе.
Вот что вам нужно знать:
Гены, связанные с болезнью Паркинсона
Существует длинный список генов, которые, как известно, способствуют развитию болезни Паркинсона, и, возможно, еще предстоит открыть еще много генов. Вот некоторые из основных игроков:
SNCA: SNCA производит белок альфа-синуклеин.В клетках мозга людей с болезнью Паркинсона этот белок собирается в группы, называемые тельцами Леви. Мутации в гене SNCA возникают при болезни Паркинсона с ранним началом.
PARK2: Ген PARK2 вырабатывает белок паркин, который обычно помогает клеткам расщеплять и перерабатывать белки.
PARK7: Мутации в этом гене вызывают редкую форму болезни Паркинсона с ранним началом. Ген PARK7 производит белок DJ-1, который защищает от митохондриального стресса.
PINK1: Белок, производимый PINK1, представляет собой протеинкиназу, которая защищает митохондрии (структуры внутри клеток) от стресса. Мутации PINK1 возникают при раннем начале болезни Паркинсона.
LRRK2: Белок, продуцируемый LRRK2, также является протеинкиназой. Мутации в гене LRRK2 связаны с поздним началом болезни Паркинсона.
Среди наследственных случаев болезни Паркинсона характер наследования различается в зависимости от вовлеченных генов. Если задействованы гены LRRK2 или SNCA, болезнь Паркинсона, вероятно, унаследована только от одного родителя.Это называется аутосомно-доминантным паттерном , когда вам нужно изменить только одну копию гена, чтобы заболевание возникло.
Если задействован ген PARK2, PARK7 или PINK1, он обычно находится в аутосомно-рецессивном паттерне , когда вам нужно изменить две копии гена, чтобы заболевание возникло. Это означает, что две копии гена в каждой клетке были изменены. Оба родителя передали измененный ген, но сами могли не иметь никаких признаков болезни Паркинсона.
Исследование генов и болезни Паркинсона
«Сейчас наша основная задача — понять, как мутации в этих генах вызывают болезнь Паркинсона», — говорит Доусон. Особенно интересен ген SNCA, ответственный за выработку белка, который «скапливается» в головном мозге и вызывает симптомы.
«Наше исследование пытается понять, как работает альфа-синуклеин, как он проходит через мозг», — говорит Доусон. «Согласно последней теории, он передается от ячейки к ячейке, и наша работа поддерживает эту идею.Мы определили белок, который позволяет скоплениям альфа-синуклеина проникать в клетки, и надеемся, что удастся разработать терапию, которая препятствует этому процессу ».
Болезнь Бехчета — NHS
Болезнь Бехчета или синдром Бехчета — редкое и малоизученное заболевание, которое приводит к воспалению кровеносных сосудов и тканей.
Подтверждение диагноза болезни Бехчета может быть трудным, потому что симптомы очень широкие и общие (они могут быть общими с рядом других состояний).
Симптомы болезни Бехчета
Основные симптомы болезни Бехчета включают:
- генитальные язвы и язвы во рту
- красные, болезненные глаза и помутнение зрения
- угревидные пятна
- головные боли
- болезненные, жесткие и опухшие суставы
В тяжелых случаях также существует риск серьезных и потенциально опасных для жизни проблем, таких как необратимая потеря зрения и инсульты.
Большинство людей с этим заболеванием испытывают эпизоды, когда их симптомы являются серьезными (обострения или рецидивы), за которыми следуют периоды, когда симптомы исчезают (ремиссия).
Со временем некоторые симптомы могут исчезнуть и стать менее неприятными, хотя они могут никогда не исчезнуть полностью.
Прочтите о симптомах болезни Бехчета
Диагностика болезни Бехчета
Не существует окончательного теста, который можно было бы использовать для диагностики болезни Бехчета.
Для проверки признаков заболевания или для исключения других причин может потребоваться несколько тестов, в том числе:
- анализов крови
- анализов мочи
- сканирований, таких как рентген, компьютерная томография или МРТ просканируйте
- биопсию кожи
- тест патергию, который включает прокалывание вашей кожи иглой, чтобы увидеть, появится ли определенное красное пятно в течение следующих дней или двух; люди с болезнью Бехчета часто имеют особенно чувствительную кожу
В текущих руководствах говорится, что диагноз болезни Бехчета обычно можно уверенно поставить, если за последние 12 месяцев у вас было не менее 3 эпизодов язв во рту и у вас было не менее 2 из них. следующие симптомы:
- генитальные язвы
- воспаление глаз
- кожные поражения (любые необычные новообразования или аномалии, которые развиваются на коже)
- патергия (гиперчувствительная кожа)
Перед постановкой диагноза необходимо исключить другие потенциальные причины сделан.
Причины болезни Бехчета
Причина болезни Бехчета неизвестна, хотя большинство экспертов считают, что это аутовоспалительное заболевание.
При аутовоспалительном заболевании иммунная система — естественная защита организма от инфекций и болезней — по ошибке атакует здоровые ткани.
В случае болезни Бехчета считается, что иммунная система по ошибке атакует кровеносные сосуды.
Неясно, что вызывает эту проблему с иммунной системой, но две вещи, как полагают, играют роль:
- генов — болезнь Бехчета, как правило, гораздо чаще встречается в определенных этнических группах, где гены, связанные с этим заболеванием могут быть более распространенными
- экологические факторы — хотя конкретный экологический фактор не был идентифицирован, показатели болезни Бехчета ниже у людей из этнической группы риска, проживающих за пределами своей родной страны
Болезнь Бехчета чаще встречается в Дальний Восток, Ближний Восток и страны Средиземноморья, такие как Турция и Израиль.
Считается, что это заболевание наиболее вероятно у людей средиземноморского, ближневосточного и азиатского происхождения, хотя оно может затронуть все этнические группы.
Лечение болезни Бехчета
Лекарства от болезни Бехчета нет, но часто можно контролировать симптомы с помощью лекарств, уменьшающих воспаление в пораженных частях тела.
Эти лекарства включают:
- стероиды — мощные противовоспалительные препараты
- иммунодепрессанты — лекарства, снижающие активность иммунной системы
- биологические методы лечения — лекарства, которые воздействуют на биологические процессы, участвующие в процессе воспаления
Ваш Медицинская бригада разработает для вас конкретный план лечения в зависимости от ваших симптомов.
Прочтите о лечении болезни Бехчета
Специализированные центры болезни Бехчета
В Англии созданы 3 центра передового опыта NHS, которые помогают диагностировать и лечить людей с болезнью Бехчета.
Они расположены в Лондоне, Бирмингеме и Ливерпуле.
Вас могут направить в один из этих центров для подтверждения диагноза. Персонал этих центров также может поддерживать связь со специалистами в других центрах, чтобы помочь в ведении пациента и лечении, даже если их не принимают напрямую.
Дополнительную информацию об этих центрах можно найти на веб-сайте центров передового опыта по синдрому Бехчета.
Если у вас болезнь Бехчета, ваша клиническая бригада передаст информацию о вас в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Дополнительная информация о болезни Бехчета
Естественный ответ на получение диагноза сложного состояния, такого как болезнь Бехчета, — это узнать как можно больше об этом заболевании.
Однако в Великобритании это может быть сложно, потому что болезнь Бехчета настолько редка, что многие медицинские работники мало о ней знают.
Хорошее место, чтобы узнать больше о болезни Бехчета, можно найти в Великобритании Бехчета — главной британской группе поддержки пациентов с болезнью Бехчета.
На его веб-сайте есть информация о различных аспектах болезни Бехчета, форум, блоги и ссылки на другие полезные ресурсы.
Последняя проверка страницы: 20 ноября 2019 г.
Срок следующего рассмотрения: 20 ноября 2022 г.
Бывают ли сердечные заболевания в вашей семье?
Найдите время, чтобы собрать информацию о сердечных заболеваниях в вашей семье, и поделитесь этой информацией со своим врачом и другими членами семьи.Ваш врач может помочь вам принять меры, чтобы снизить вероятность заболевания сердца.
Соберите и поделитесь историей болезни сердца вашей семьи
Ежегодно в США около 655 000 человек умирают от болезней сердца. Некоторые медицинские условия, такие как высокий уровень холестерина, высокое кровяное давление и диабет, а также факторы образа жизни, такие как нездоровая диета, недостаток физической активности и курение, могут повысить вероятность развития сердечных заболеваний. Кроме того, наличие близких кровных родственников с сердечными заболеваниями может повысить вероятность сердечного заболевания.
Если у вас есть семейная история болезни сердца, соберите информацию о ваших родственниках с сердечным заболеванием, в том числе о том, в каком возрасте им был поставлен диагноз. Это особенно важно, если у вас есть родитель, брат или сестра, страдающие сердечным заболеванием. Поделитесь этой информацией со своим врачом, чтобы вы вместе работали над тем, как снизить вероятность сердечного заболевания.
Эти шаги могут включать
- здоровое питание,
- человек физически активны,
- поддержание здорового веса,
- не курить,
- ограничение употребления алкоголя,
- проверка холестерина,
- контроль артериального давления,
- управление диабетом, если он у вас есть,
- прошли скрининговые тесты, рекомендованные вашим врачом, и
- принимать лекарства, если необходимо для лечения высокого уровня холестерина, высокого кровяного давления или диабета.
человек
Рианнон пишет о семейной гиперхолестеринемии.
Важность семейного анамнеза: история Рианнон
У отца Рианнон был свой первый сердечный приступ в 36 лет, и он умер от одного из них в 51 год. Она думала, что его болезнь сердца возникла из-за его курения и нездоровой диеты, пока она не проверила уровень холестерина в 30 лет. Ее липопротеины низкой плотности (ЛПНП) ) уровень холестерина был почти в три раза выше, чем ожидалось для женщины ее возраста.«Мне никогда не проверяли уровень холестерина, потому что я всегда слышал, что диеты и физических упражнений достаточно, чтобы справиться с этим. Это просто не должно было быть проблемой для молодых, активных, не курящих и не страдающих лишним весом. Верно? Неправильный. И, мальчик, я был неправ ».
У некоторых людей, например у Рианнон, есть общее генетическое заболевание, называемое семейной гиперхолестеринемией (СГ). У людей с СГ повышен уровень холестерина ЛПНП, что увеличивает вероятность развития сердечных заболеваний в более молодом возрасте и увеличивает риск смерти от этого заболевания.Для многих людей с СГ одной диеты и физических упражнений недостаточно для контроля уровня холестерина, и им требуются лекарства, такие как статины.
После того, как Рианнон узнала, что у нее СГ, она сначала забеспокоилась: «У меня случится сердечный приступ, и я умру в молодом возрасте, как мой отец?» Тем не менее, она работала со своим врачом, чтобы предпринять шаги по снижению уровня ЛПНП. «Я научился управлять своим холестерином с помощью лекарств, диеты и физических упражнений. Я регулярно проверяю свою кровь [холестерин] … Во время моего последнего осмотра мои показатели холестерина были лучшими из когда-либо существовавших.”
Информация о том, что у человека есть СГ, помогает не только этому человеку, но и всей его семье. Другие члены семьи могут быть проверены на СГ, и люди с этим расстройством могут предпринять шаги, чтобы снизить свои шансы на развитие и смерть от сердечных заболеваний. Как объясняет Рианнон: «Мне очень повезло, что я узнала о своей СГ до того, как со мной случилось что-то серьезное, и я надеюсь избежать сердечных заболеваний с помощью профилактики».
болезнь | Определение, типы и контроль
Болезнь , любое вредное отклонение от нормального структурного или функционального состояния организма, обычно связанное с определенными признаками и симптомами и отличающееся по своей природе от физического повреждения.Больной организм обычно проявляет признаки или симптомы, указывающие на его ненормальное состояние. Таким образом, необходимо понимать нормальное состояние организма, чтобы распознать признаки болезни. Тем не менее, резкая граница между болезнью и здоровьем не всегда очевидна.
Землетрясение на Гаити в 2010 году: холера
Ожидающие лечения гаитяне, страдающие симптомами холеры, получают помощь от других жителей, Сент-Марк, Гаити, октябрь 2010 года.
Ramon Espinosa / AP
Британская викторина
Медицинские термины и викторина для первопроходцев
Кто открыл основные группы крови? Что вызывает заболевание крови талассемией? Проверьте, что вы знаете о медицине, пройдя этот тест.
Исследование болезни называется патологией. Он включает определение причины (этиологии) заболевания, понимание механизмов его развития (патогенеза), структурных изменений, связанных с болезненным процессом (морфологические изменения), и функциональных последствий этих изменений. Правильное определение причины заболевания необходимо для определения правильного курса лечения.
Люди, другие животные и растения восприимчивы к тем или иным заболеваниям.Однако то, что нарушает нормальное функционирование одного типа организмов, может не влиять на другие типы.
Основные отличия
Нормальное состояние организма представляет собой состояние тонкого физиологического баланса или гомеостаза с точки зрения химических, физических и функциональных процессов, поддерживаемого комплексом механизмов, которые до конца не изучены. Таким образом, в фундаментальном смысле болезнь представляет собой последствия нарушения механизмов гомеостатического контроля.В некоторых случаях пораженные механизмы четко обозначены, но в большинстве случаев комплекс механизмов нарушается, изначально или последовательно, и точное определение патогенеза последующего заболевания невозможно. Например, смерть у людей и других млекопитающих часто является прямым следствием сердечной или легочной недостаточности, но предшествующая последовательность событий может быть очень сложной, включая нарушения других систем органов и нарушение других механизмов контроля.
Получите подписку Britannica Premium и получите доступ к эксклюзивному контенту.Подпишитесь сейчас
Первоначальная причина болезненного состояния может лежать в самом отдельном организме, и тогда болезнь называется идиопатической, врожденной, первичной или «существенной». Это может быть следствием курса лечения либо как неизбежный побочный эффект, либо потому, что само лечение было опрометчивым; в любом случае болезнь классифицируется как ятрогенная. Наконец, болезнь может быть вызвана каким-либо внешним по отношению к организму агентом, например, химическим веществом, которое является токсичным агентом. В этом случае болезнь неинфекционная; то есть он влияет только на индивидуальный организм, подверженный ему.Внешний агент может сам по себе быть живым организмом, способным размножаться внутри хозяина и впоследствии инфицировать другие организмы; в этом случае болезнь считается инфекционной.
Неинфекционные заболевания
Неинфекционные болезни обычно носят длительный характер и медленно прогрессируют, поэтому их иногда также называют хроническими заболеваниями. Они могут возникать в результате воздействия окружающей среды или генетически обусловленных аномалий, которые могут проявляться при рождении или проявляться позже в жизни.Всемирная организация здравоохранения (ВОЗ) определила четыре основных типа неинфекционных заболеваний: рак, сердечно-сосудистые заболевания (например, сердечный приступ, инсульт), хронические респираторные заболевания (например, астма) и сахарный диабет. По оценкам ВОЗ, в совокупности на эти четыре группы состояний приходится 82 процента всех случаев смерти от неинфекционных заболеваний.
рак головного мозга; магнитно-резонансная томография (МРТ)
Изображение человеческого мозга, пораженного раком, полученное с помощью магнитно-резонансной томографии (МРТ).Ярко-синяя область указывает на то, что рак распространился на затылочную долю (внизу справа).
© Photodisc / Thinkstock
Неинфекционные заболевания, возникающие в результате наследственных генетических аномалий, часто лишают человека возможности выжить без какой-либо формы лечения. Примеры наследственных заболеваний включают муковисцидоз, синдром Дауна и врожденные нарушения обмена веществ, которые присутствуют при рождении. Примеры наследственных заболеваний, которые возникают во взрослом возрасте, включают болезнь Хантингтона и определенные формы рака (например,g., семейный рак молочной железы, включающий наследственные мутации в любом из генов BRCA1 или BRCA2 ).
Болезнь движения: симптомы и лечение
Обзор
Что такое укачивание?
Укачивание возникает, когда ваш мозг не может воспринимать информацию, поступающую из ваших глаз, ушей и тела.Постоянное движение — в машине, самолете, лодке или даже в парке развлечений — может вызвать тошноту, липкость или тошноту в животе. Некоторых людей рвет. Укачивание, морская болезнь или воздушная болезнь — это укачивание.
Кто может заболеть укачиванием?
По оценкам, каждый третий человек в какой-то момент заболевает укачиванием. Наибольшему риску подвержены женщины и дети в возрасте от 2 до 12 лет. Тем не менее, это состояние может повлиять на кого угодно.
Эти факторы увеличивают ваши шансы заболеть укачиванием:
Симптомы и причины
Что вызывает укачивание?
Ваш мозг получает сигналы от чувствительных к движению частей вашего тела: глаз, внутреннего уха, мышц и суставов.Когда эти части отправляют противоречивую информацию, ваш мозг не знает, неподвижны вы или движетесь. Беспорядочная реакция вашего мозга вызывает тошноту.
Например, при езде в автомобиле ваш:
- Глаза видят проезжающие деревья и регистрируют движение.
- Внутреннее ухо ощущает движение.
- Мышцы и суставы чувствуют, что ваше тело неподвижно.
- Мозг чувствует разрыв между этими сообщениями.
Многие действия могут вызвать укачивание, например:
- Аттракционы и виртуальная реальность.
- Чтение в движении.
- Поездка на лодке, автомобиле, автобусе, поезде или самолете.
- Видеоигры и фильмы.
Каковы симптомы укачивания?
Морская болезнь может застать вас врасплох. В один момент вы можете почувствовать себя хорошо, а затем внезапно почувствовать некоторые из этих симптомов:
Диагностика и тесты
Как диагностируется укачивание?
Ваш лечащий врач попросит вас описать ваши симптомы и то, что вызывает эти симптомы.Ваш лечащий врач также может провести медицинский осмотр и проверить ваши глаза и уши.
Ведение и лечение
Как лечить или лечить укачивание?
У вас есть несколько вариантов предотвращения укачивания или лечения симптомов. Лечение укачивания включает:
- Антигистаминные препараты: Антигистаминные препараты, обычно используемые для лечения аллергии, также могут предотвратить укачивание и облегчить симптомы.Эффективны только антигистаминные препараты, вызывающие сонливость. Формулы, предотвращающие сонливость, не помогут.
- Пластыри: Пластыри для кожи Scopolamine (Transderm Scop®) или таблетки для приема внутрь предотвращают тошноту и рвоту. Вы прикрепляете пластырь за ухом как минимум за четыре часа до поездки. Через три дня вы снимаете пластырь и накладываете новый. Это лекарство может вызвать сухость во рту и разрешено только для взрослых.
Каковы осложнения укачивания?
Укачивание не вызывает серьезных проблем.В редких случаях у некоторых людей не прекращается рвота. Чрезмерная рвота может вызвать обезвоживание и низкое кровяное давление (гипотензию).
Профилактика
Как предотвратить укачивание?
Эти действия могут снизить ваши шансы заболеть или облегчить симптомы, если они возникнут:
- Травы: Вдохните успокаивающий аромат мяты, имбиря или лаванды.Пососите леденцы из перечной мяты или имбиря.
- Диета и питье: Пейте много воды. Перед путешествием выбирайте нежирные, безвкусные и крахмалистые продукты. Избегайте тяжелой пищи и жирной, острой или кислой пищи, которая может вызвать расстройство желудка. Не употребляйте алкоголь и не курите.
- Свежий воздух: Направляйте вентиляционные отверстия на вас. И опускать окна в машинах.
- Взгляд вдаль: Положите телефон, планшет или книгу. Вместо этого смотрите на объект вдалеке или на горизонте.
- Откиньтесь на спинку кресла: Откиньтесь, если возможно, и закройте глаза.
- Точки давления: Носите браслеты для акупрессуры.
Во время путешествия вы всегда должны смотреть вперед. Место, где вы сидите, также может иметь значение, чтобы свести к минимуму мешающие движения:
- Лодка: Сядьте в середине лодки на верхней палубе.
- Автобус: Выберите место у окна.
- Автомобиль: Сядьте на переднее пассажирское сиденье.
- Круизное судно: Забронируйте каюту в передней или средней части судна. Если можете, выберите один на более низком уровне, ближе к воде.
- Самолет: Сядьте в секцию крыла.
- Поезд: Выберите сиденье у окна, обращенное вперед.
Перспективы / Прогноз
Каков прогноз (перспективы) для людей, страдающих укачиванием?
Морская болезнь может сделать путешествие стрессовым и неприятным.Но симптомы должны исчезнуть, когда вы перестанете двигаться.
Жить с
Когда мне следует позвонить поставщику медицинских услуг?
Вы должны позвонить своему врачу, если у вас возникли проблемы:
- Хроническая постоянная тошнота или рвота.
- Симптомы укачивания, когда вы не двигаетесь.
- Признаки обезвоживания.
Какие вопросы я должен задать своему врачу?
Вы можете спросить своего поставщика медицинских услуг:
- Как предотвратить укачивание?
- Как лучше всего лечить укачивание?
- Каковы побочные эффекты лечения?
Записка из клиники Кливленда
Почти каждый в какой-то момент чувствует укачивание. Рвота может вызвать тошноту и тошноту.Вы не всегда можете избежать движения, от которого вас тошнит, особенно во время путешествий. Если вы склонны к укачиванию, поговорите со своим врачом о способах предотвращения заболевания и о том, что делать, если вы заболели. Вы можете предпринять шаги, чтобы получить больше удовольствия от путешествия, не прибегая к сумке для барбекю.
Хроническая болезнь почек: симптомы, причины и лечение
Хроническая болезнь почек — это медленная и прогрессирующая потеря функции почек в течение нескольких лет.В конце концов, у человека разовьется необратимая почечная недостаточность.
Хроническая болезнь почек, также известная как хроническая почечная недостаточность, хроническая почечная недостаточность или хроническая почечная недостаточность, гораздо более распространена, чем люди думают; он часто остается незамеченным и недиагностированным до тех пор, пока болезнь не перейдет в серьезную стадию.
Люди нередко осознают, что у них хроническая почечная недостаточность, только тогда, когда их функция почек снижается до 25 процентов от нормы.
По мере развития почечной недостаточности и серьезного нарушения функции органа в организме может быстро накапливаться опасный уровень отходов и жидкости.Лечение направлено на остановку или замедление прогрессирования заболевания — обычно это делается путем устранения его первопричины.
Поделиться на Pinterest Хроническая болезнь почек редко проявляется симптомами до поздних стадий, поэтому тем, кто подвержен риску, рекомендуется обследование.
Хроническая почечная недостаточность, в отличие от острой почечной недостаточности, является медленным и постепенно прогрессирующим заболеванием. Даже если одна почка перестает функционировать, другая может выполнять нормальные функции. Обычно признаки и симптомы становятся заметными только тогда, когда болезнь достаточно далеко продвинулась и состояние не стало тяжелым; к этому времени большая часть повреждений необратима.
Важно, чтобы у людей с высоким риском развития почечной недостаточности регулярно проверялась функция почек. Раннее обнаружение может значительно помочь предотвратить серьезное повреждение почек.
Наиболее частые признаки и симптомы хронического заболевания почек включают:
- анемию
- кровь в моче
- темная моча
- снижение умственной активности
- снижение диуреза
- отек — опухшие ступни, руки и лодыжки (лицо при сильном отеке)
- утомляемость (усталость)
- гипертония (высокое кровяное давление)
- бессонница
- кожный зуд, может стать постоянным
- потеря аппетита
- мужская неспособность достичь или поддерживать эрекцию (эректильная дисфункция)
- более частое мочеиспускание, особенно ночью
- мышечные спазмы
- мышечные судороги
- тошнота
- боль в боку или в средней или нижней части спины
- одышка (одышка)
- белок в моче
- резкое изменение массы тела
- необъяснимые головные боли
Изменения скорости клубочковой фильтрации позволяют оценить, насколько далеко зашло заболевание почек. является.В Великобритании и многих других странах стадии заболевания почек классифицируются следующим образом:
Стадия 1 — СКФ в норме. Однако признаков заболевания почек обнаружено не было.
Стадия 2 — СКФ ниже 90 миллилитров, обнаружены признаки заболевания почек.
Стадия 3 — СКФ ниже 60 миллилитров, независимо от того, были ли обнаружены признаки заболевания почек.
Стадия 4 — уровень GRF ниже 30 миллилитров, независимо от того, были ли обнаружены признаки заболевания почек.
Стадия 5 — СКФ ниже 15 миллилитров. Произошла почечная недостаточность.
Большинство пациентов с хронической болезнью почек редко прогрессируют после стадии 2. Важно, чтобы заболевание почек было диагностировано и началось лечение на ранней стадии, чтобы предотвратить серьезное повреждение.
Пациентам с диабетом следует ежегодно сдавать анализ на микроальбуминурию (небольшое количество белка) в моче. Этот тест может выявить раннюю диабетическую нефропатию (раннее повреждение почек, связанное с диабетом).
В настоящее время не существует лекарства от хронической болезни почек. Однако некоторые методы лечения могут помочь контролировать признаки и симптомы, снизить риск осложнений и замедлить прогрессирование заболевания.
Пациентам с хроническим заболеванием почек обычно необходимо принимать большое количество лекарств. Лечение включает:
Лечение анемии
Гемоглобин — это вещество в красных кровяных тельцах, которое переносит жизненно важный кислород по всему телу. Если уровень гемоглобина низкий, у пациента анемия.
Некоторым пациентам с заболеванием почек, страдающим анемией, требуется переливание крови. Пациенту с заболеванием почек обычно приходится принимать добавки железа либо в форме ежедневных таблеток сульфата железа, либо иногда в виде инъекций.
Баланс фосфатов
Люди с заболеванием почек не могут должным образом выводить фосфаты из своего организма. Пациентам будет рекомендовано снизить потребление пищевых фосфатов — обычно это означает сокращение потребления молочных продуктов, красного мяса, яиц и рыбы.
Высокое кровяное давление
Высокое кровяное давление — обычная проблема для пациентов с хронической болезнью почек. Важно снизить артериальное давление, чтобы защитить почки и, следовательно, замедлить прогрессирование заболевания.
Кожный зуд
Антигистаминные препараты, такие как хлорфенамин, могут помочь облегчить симптомы зуда.
Лекарства от болезней
Если токсины накапливаются в организме из-за того, что почки не работают должным образом, пациенты могут чувствовать себя плохо (тошнота).Лекарства, такие как циклизин или метаклопрамид, помогают облегчить недомогание.
НПВП (нестероидные противовоспалительные препараты)
НПВП, такие как аспирин или ибупрофен, следует избегать и принимать только по рекомендации врача.
Завершающая стадия лечения
Это когда почки функционируют менее чем на 10-15 процентов от нормальной производительности. Меры, которые использовались до сих пор — диета, лекарства и методы лечения, контролирующие основные причины, — уже недостаточны. Почки пациентов с терминальной стадией заболевания почек не могут справиться с процессом удаления отходов и жидкости самостоятельно — пациенту потребуется диализ или трансплантация почки, чтобы выжить.
Большинство врачей стараются отложить необходимость диализа или трансплантации почки как можно дольше, потому что они несут в себе риск потенциально серьезных осложнений.
Диализ почек
Существует два основных типа диализа почек. У каждого типа также есть подтипы. Два основных типа:
Гемодиализ : кровь откачивается из тела пациента и проходит через диализатор (искусственная почка). Примерно трижды в неделю пациенту проводится гемодиализ.Каждый сеанс длится не менее 3 часов.
Сейчас эксперты признают, что более частые сеансы улучшают качество жизни пациента, но современные домашние диализные аппараты делают возможным более регулярное использование гемодиализа.
Перитонеальный диализ: Кровь фильтруется в брюшной полости пациента; в брюшной полости, которая содержит обширную сеть крошечных кровеносных сосудов. В брюшную полость имплантируется катетер, в который вводится и сливается диализный раствор до тех пор, пока это необходимо для удаления отходов и лишней жидкости.
Трансплантат почки
Донор и реципиент почки должны иметь одинаковую группу крови, белки клеточной поверхности и антитела, чтобы свести к минимуму риск отторжения новой почки. Братья и сестры или очень близкие родственники обычно являются лучшими донорами. Если живой донор невозможен, начнутся поиски донора трупа (мертвого человека).
Соблюдение правильной диеты жизненно важно для эффективного лечения почечной недостаточности. Ограничение количества белка в рационе может помочь замедлить прогрессирование заболевания.
Диета также может помочь облегчить симптомы тошноты.
Для контроля гипертонии необходимо тщательно регулировать потребление соли. Потребление калия и фосфора со временем также может потребоваться ограничить.
Витамин D
Пациенты с заболеванием почек обычно имеют низкий уровень витамина D. Витамин D необходим для здоровья костей. Витамин D, который мы получаем от солнца или пищи, должен быть активирован почками, прежде чем организм сможет его использовать. Пациентам можно назначить альфакальцидол или кальцитриол.
Задержка жидкости
Людям с хроническим заболеванием почек необходимо соблюдать осторожность при приеме жидкости. Большинству пациентов будет предложено ограничить потребление жидкости. Если почки не работают должным образом, пациент гораздо более подвержен накоплению жидкости.
Почки осуществляют сложную систему фильтрации в нашем организме — лишние отходы и жидкие вещества удаляются из крови и выводятся из организма.
В большинстве случаев почки могут выводить большинство отходов, производимых нашим организмом.Однако, если приток крови к почкам нарушен, они не работают должным образом из-за повреждения или болезни, или если отток мочи затруднен, могут возникнуть проблемы.
В большинстве случаев прогрессирующее поражение почек является результатом хронического заболевания (длительного заболевания), например:
- Диабет — хроническое заболевание почек связано с диабетом 1 и 2 типа. Если у пациента диабет не контролируется должным образом, избыток сахара (глюкозы) может накапливаться в крови. Заболевания почек не распространены в течение первых 10 лет диабета; чаще это происходит через 15-25 лет после установления диагноза диабета.
- Гипертония (высокое кровяное давление) — высокое кровяное давление может повредить клубочки — части почек, участвующие в фильтрации продуктов жизнедеятельности.
- Затрудненный отток мочи — если отток мочи заблокирован, он может вернуться обратно в почки из мочевого пузыря (пузырно-мочеточниковый рефлюкс). Блокированный поток мочи увеличивает давление на почки и нарушает их функцию. Возможные причины включают увеличенную простату, камни в почках или опухоль.
- Болезни почек — включая поликистоз почек, пиелонефрит или гломерулонефрит.
- Стеноз почечной артерии — почечная артерия сужается или блокируется до того, как войдет в почку.
- Определенные токсины — включая топливо, растворители (например, четыреххлористый углерод) и свинец (а также краски на основе свинца, трубы и материалы для пайки). Даже некоторые украшения содержат токсины, которые могут привести к хронической почечной недостаточности.
- Проблема развития плода — если почки не развиваются должным образом у будущего ребенка, пока он развивается в утробе матери.
- Системная красная волчанка — аутоиммунное заболевание. Собственная иммунная система организма атакует почки, как если бы они были чужеродной тканью.
- Малярия и желтая лихорадка — известно, что они вызывают нарушение функции почек.
- Некоторые лекарства — чрезмерное употребление, например, НПВП (нестероидных противовоспалительных средств), таких как аспирин или ибупрофен.
- Незаконное употребление психоактивных веществ — таких как героин или кокаин.
- Травма — резкий удар или физическое повреждение почки (ей).
Факторы риска
Следующие состояния или ситуации связаны с более высоким риском развития заболевания почек:
- семейный анамнез заболевания почек
- возраст — хроническое заболевание почек гораздо чаще встречается среди людей старше 60 лет
- атеросклероз
- непроходимость мочевого пузыря
- хронический гломерулонефрит
- врожденное заболевание почек (заболевание почек, проявляющееся при рождении)
- диабет — один из наиболее распространенных факторов риска
- гипертония
- красная волчанка
- чрезмерное воздействие некоторых токсинов на клетки болезнь
- некоторые лекарства
Врач проверит признаки и расспросит пациента о симптомах.Также можно заказать следующие анализы:
- Анализ крови — можно заказать анализ крови, чтобы определить, адекватно ли отфильтровываются отходы. Если уровни мочевины и креатинина постоянно высоки, врач, скорее всего, диагностирует терминальную стадию заболевания почек.
- Анализ мочи — анализ мочи помогает определить, есть ли в моче кровь или белок.
- Сканирование почек — Сканирование почек может включать магнитно-резонансную томографию (МРТ), компьютерную томографию (КТ) или ультразвуковое сканирование.Цель состоит в том, чтобы определить, есть ли какие-либо препятствия для оттока мочи. Это сканирование также может выявить размер и форму почек — на поздних стадиях заболевания почек почки меньше и имеют неровную форму.
- Биопсия почки — извлекается небольшой образец ткани почек и исследуется на предмет повреждения клеток. Анализ почечной ткани помогает поставить точный диагноз заболевания почек.
- Рентгеновский снимок грудной клетки — здесь цель состоит в том, чтобы проверить наличие отека легких (задержка жидкости в легких).
- Скорость клубочковой фильтрации (СКФ) — СКФ — это тест, который измеряет скорость клубочковой фильтрации — он сравнивает уровни продуктов жизнедеятельности в крови и моче пациента. СКФ определяет, сколько миллилитров отходов почки могут отфильтровать в минуту. Почки здоровых людей обычно могут фильтровать более 90 мл в минуту.
Если хроническая болезнь почек прогрессирует до почечной недостаточности, возможны следующие осложнения:
- анемия
- поражение центральной нервной системы
- сухая кожа или изменение цвета кожи
- задержка жидкости
- гиперкалиемия, когда уровень калия в крови повышается может привести к повреждению сердца
- бессонница
- нижнее половое влечение
- мужская эректильная дисфункция
- остеомаляция, когда кости становятся слабыми и легко ломаются
- перикардит, когда мешкообразная оболочка вокруг сердца воспаляется
- язвы желудка
- слабая иммунная система
Управление хроническим заболеванием
Некоторые состояния повышают риск хронического заболевания почек (например, диабета).